Medicinsk expert av artikeln

Nya publikationer
hamartom
Senast recenserade: 29.06.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
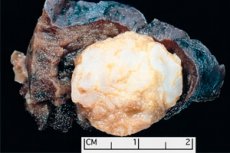
Tumörliknande formationer lokaliserade i någon anatomisk region till följd av onormal tillväxt av godartad vävnad definieras inom medicin som hamartom (från grekiska hamartia - fel, defekt). [ 1 ]
Epidemiologi
Statistiskt sett står hamartom för 1,2 % av alla godartade tumörer. Förekomsten av pulmonella hamartom uppskattas till cirka 0,25 % av den allmänna befolkningen och står för upp till 8 % av alla pulmonella tumörer. De flesta pulmonella hamartom diagnostiseras av en slump hos patienter i åldern 40 till 70 år, men är mycket sällsynta inom pediatrisk praxis.
Generellt sett diagnostiseras de flesta hamartom hos män, även om de i njuren är vanligare hos kvinnor och identifieras i medelåldern.
Cirka 5% av godartade brösttumörer är hamartom, och de drabbar oftast kvinnor över 35 år.
80–90 % av hamartomatösa lesioner i hjärnan och mer än 50 % av hamartom i hjärtat är associerade med tuberös skleros.
Orsaker hamartom
Gamartom tillhör medfödda missbildningar och är formationer av godartad karaktär, som bildas från mesenkymala vävnader som härrör från germinala skikt. Och orsakerna till deras uppkomst är förknippade med okontrollerad celldelning av cytologiskt normala vävnader (bindvävnad, glatt muskulatur, fett eller brosk), karakteristiska för en given anatomisk plats, och deras fokala överväxt under embryogenesen av nästan vilket organ eller anatomisk struktur som helst.
Förekomsten av flera hamartom hos samma patient kallas ofta hamartomatos eller pleiotropiskt hamartom.
Dessa tumörer kan förekomma sporadiskt eller i närvaro av vissa autosomalt dominanta ärftliga sjukdomar såväl som genetiskt betingade syndrom.
I många fall bildas hamartom när en sällsynt genetisk sjukdom av multisystemisk natur – tuberös skleros – manifesterar sig kort efter födseln, eller vid Recklinghausens familjära sjukdom – neurofibromatos typ 1. [ 2 ]
Riskfaktorer
De viktigaste riskfaktorerna för hamartombildning inkluderar förekomsten av så kallade genetiska syndrom av hamartomatös polypos i patientens historia, inklusive:
- Multipelt hamartomsyndrom - Cowden syndrom, där flera hamartom av ekto-, ento- och mesodermalt ursprung bildas, gastrointestinal polypos och mukokutana manifestationer observeras;
- Peutz-Jeghers-Turen syndrom (kännetecknas av utveckling av godartade hamartomatösa polyper i mag-tarmkanalen);
- Proteus syndrom;
- Weils syndrom - juvenil polypos i tjocktarmen;
- Bannayan-Riley-Ruvalcabas syndrom, som liksom Cowdens syndrom producerar flera hamartom (hamartomatösa polyper) i tarmen;
- Carney-Stratakis syndrom och Carney-komplex.
Dessutom bildas hamartom hos patienter med ärftligt Watsons syndrom och i fall av sporadiskt förekommande eller kongenitalt Pallister-Halls syndrom med hypotalamiskt hamartom och polydaktyli.
Patogenes
Mekanismen för ökad proliferation av germinalvävnader med bildandet av tumörliknande missbildningar i olika organ förklaras av kromosomavvikelser och genmutationer som kan uppstå spontant eller ärvas.
Vid tuberös skleros har man identifierat mutationer i TSC1- eller TSC2-generna – tumörsuppressorer som förhindrar och hämmar överdriven proliferation – för snabb eller okontrollerad celltillväxt och delning. Och vid neurofibromatos typ 1 och Watsons syndrom – mutationer i könscellerna i den mitokondriella tumörsuppressorgenen NF1.
Vid hamartomtumörsyndrom, som kombinerar Cowden-, Protea-, Bannayan-Riley-Ruvalcaba- och juvenil polypossyndrom, är patogenesen associerad med mutation av PTEN-genen, som kodar för ett enzym som är involverat i regleringen av proliferation och anses vara en tumörsuppressorgen.
Mutationer i STK11-genen som kodar för strukturen och funktionen hos ett av de transmembrana serinenzymerna, vilket minskar dess förmåga att hämma celldelning, leder till Peutz-Jeghers-Turen syndrom, med utveckling av tarmpolyper och pigmenterade hudlesioner. En mutation i GLI3-genen, en transkriptionsfaktor involverad i intrauterin vävnadsbildning, har identifierats vid Pallister-Hall syndrom.
Således leder okontrollerad celltillväxt på grund av genmutationer till hamartombildning.
Symtom hamartom
Beroende på lokaliseringen av hamartom utmärks deras typer, och var och en av dem har sin egen struktur och symtomatologi.
Hamartom i lungan
Pulmonellt hamartom kan bildas i vilken lob som helst och i perifera delar av lungorna och består av normal vävnad som finns i lungorna: fettvävnad, epitelvävnad, fibrös vävnad och broskvävnad. I 80 % av fallen dominerar kondroidkomponenten (hyalinbroskceller) med inkludering av adipocyter - fettvävnadsceller och luftvägsepitelceller. [ 3 ]
De tidigare namnen: kondroid hamartom, mesenkymom, kondromatöst hamartom eller hamartokondrom rekommenderas för närvarande inte av WHO.
Mesenkymalt cystisk hamartom i lungan är däremot mindre vanligt och är associerat med Cowdens syndrom hos de flesta patienter.
Hamartomatös lesion i lungan kan inte manifestera sig själv, men kan orsaka symtom i form av kronisk hosta (ofta med hemoptys), väsande andning vid andning och andningssvårigheter. [ 4 ]
Ett hamartom i hjärtat
Godartade primära hjärttumörer hos vuxna inkluderar moget myocythamartom, och hos spädbarn och barn med tuberös skleros, rabdomyom, det vill säga myokardiellt hamartom i kamrarna eller interventrikulärt septum. [ 5 ]
Moget kardiomyocythamartom utvecklas i kammarväggen (och sällan i förmaken) och kan uppstå som multipla lesioner som är täta massor nära förknippade med det underliggande myokardiet. Tumören kan orsaka symtom på hjärtsvikt: bröstsmärta, hjärtklappning och arytmier, hjärtmumling, ödem, dyspné, cyanos.
Hjärtliga rabdomyomer, av vilka de flesta diagnostiseras under det första levnadsåret, består av hjärtmuskelvävnad som bildas av embryonala myoblaster och ser ut som fasta fokalmassor utan kapsel.
Vanligtvis uppträder dessa hamartom asymptomatiska och försvinner spontant före 4 års ålder.
Hamartomatösa lesioner anses också av vissa experter vara associerade med Carney-komplexets myxom i hjärtat. [ 6 ]
Gamartom i mag-tarmkanalen
Gastriskt hamartom är en mesenkymal massa i form av en epitelial hyperplastisk polyp i magsäcken, Peutz-Jeghers polyp och ett sällsynt myoepitelialt hamartom - med hypertrofierade glatta muskelbuntar. Andra namn för detta hamartom inkluderar myoglandulärt hamartom, adenomyomatöst hamartom och gastriskt adenomyom. Typiska kliniska manifestationer inkluderar dyspepsi, epigastrisk smärta och blödning i övre magtarmkanalen. [ 7 ], [ 8 ]
Mer information i materialet - magpolypos
Ett intestinalt hamartom är en hamartomatös eller hyperplastisk polyp i tjocktarmen, diagnostiserad som ett adenomatöst eller tubulärt adenom. När hamartomet är lokaliserat i Brunnerkörteln i tolvfingertarmen, manifesteras symtomen av smärta i epigastriet; illamående, kräkningar och gastrointestinal gastrointestinal region; illamående, kräkningar och gastrointestinal blödning (vilket indikerar tarmobstruktion); och, om den är av betydande storlek, gastrointestinal blödning. Vid myoepitelial hamartom i ileum klagar patienterna på buksmärtor, minskad kroppsvikt och utvecklar kronisk anemi. [ 9 ], [ 10 ]
Läs också - rektala polyper
Retrorektalt hamartom är ett cystiskt hamartom eller en flerkammarcysta i det retrorektala utrymmet (den lösa bindväven mellan ändtarmen och dess egen fascia) som oftast förekommer hos medelålders kvinnor. Det ser ut som en cysta som buktar ut ur ändtarmens bakre vägg, vilken är beklädd med epitel och innehåller kaotiskt arrangerade glatta muskelfibrer. Detta hamartom uppvisar smärta i nedre delen av buken och återkommande förstoppning. [ 11 ], [ 12 ]
Hamartom i levern och mjälten
Multipelt biliärt hamartom i levern är ett hamartom i de interdollika intrahepatiska gallgångarna associerat med missbildningar i deras utveckling under embryonalperioden. Detta hamartom (enkelt eller multipelt) består av slumpmässigt vidgade kluster av gallgångar och fibrokollagenöst stroma. [ 13 ]
Biliära hamartom är asymptomatiska och upptäcks vanligtvis av en slump (vid radiologisk undersökning eller laparotomi). [ 14 ]
En sällsynt och ofta av en slump upptäckt primär tumör av godartad karaktär är ett hamartom i mjälten, som består av delar av mjältens röda pulpa - i form av en väldefinierad homogen massa med fast konsistens. Denna missbildning kan vara enstaka eller flera; vid klämning av mjältparenkymet kan det uppstå en känsla av obehag och smärta i vänster subkostalområde. [ 15 ], [ 16 ]
Njurhamartom
Det vanligaste hamartomet i njuren diagnostiseras som angiomyolipoma i njuren, eftersom denna godartade tumör består av mogen fettvävnad med inbäddade glatta muskelfibrer och blodkärl. Den bildas vid tuberös skleros i 40–80 % av fallen. Att öka hamartomets storlek (mer än 4–5 cm) leder till smärta och blod i urinen. [ 17 ], [ 18 ]
Hamartom i bröstet
De WHO-accepterade diagnostiska definitionerna av brösthamartom är termer som adenolipoma, kondrolipoma och myoidhamartom. Även om mammologer ofta kallar det fibroadenolipoma, eftersom tumörformationen innehåller celler av fibrös, körtel- och fettvävnad inneslutna i en tunn bindvävskapsel med tydliga konturer. Fokala förkalkningar kan observeras vid visualisering. I detta fall saknas kliniska manifestationer. [ 19 ], [ 20 ]
Läs även - brösttumörer
Hamartom i hjärnan
En tredjedel av patienter med tuberös skleros har ett hjärnhamartom i form av intrakraniella kortikala utväxter eller tuberkler i olika lober - vid gränsen mellan grå och vit substans - eller subependymala noduler längs hjärnans kammarväggar. Astrocytiskt hamartom, ett subependymalt jättecellsastrocytom med kortikal störning, dysmorfa neuroner och stora gliaceller i hjärnparenkym (astrocyter), kan också bildas. Symtom på hjärnhamartom inkluderar anfall och utvecklingsstörning hos barn. [ 21 ], [ 22 ]
En sällsynt missbildning som uppstår under embryogenesen och är närvarande vid födseln är ett hypotalamiskt hamartom, vilket är en massa av heterotopiska neuroner och gliaceller. Allt eftersom barnets hjärna växer förstoras tumören men sprider sig inte till andra hjärnregioner. [ 23 ], [ 24 ]
Om hypertrofierad vävnad bildas i den främre delen av hypotalamus (tuber cinereum), där hypofysen fäster vid den, manifesterar missbildningen symtom på central för tidig sexuell utveckling (före 8-9 års ålder): uppkomst av akneutslag, tidig utveckling av mjölkkörtlar och tidig menarche hos flickor; tidigt könshår och röstmutation hos pojkar.
När hamartom bildas i den bakre delen av hypotalamus kan det finnas avvikelser i hjärnans elektriska aktivitet, vilket i tidig spädbarnsålder manifesteras av kramper, och i ett senare skede (åldrar 4 till 7 år) av epilepsi med fokala epileptiska anfall med plötsligt skratt eller med ofrivillig gråt, atoniska och tonisk-kloniska anfall, samt aggressionsanfall, minnes- och kognitiva problem.
Ett hypofyshamartom är ett sporadiskt förekommande godartat hypofysadenom.
Medelålders vuxna med Cowdens syndrom kan ha en sällsynt tumörliknande massa, ett hamartom i lillhjärnan, diagnostiserat som dysplastiskt cerebellärt gangliocytom eller Lhermitte-Duclos sjukdom. Symtomen kan saknas eller manifestera sig som huvudvärk, yrsel, nedsatt koordination av rörelser och förlamning av enskilda kranialnerver.
Lymfkörtelhamartom
När cellerna i glatt muskulatur och fettvävnad, såväl som blodkärl och kollagent stroma i inguinala, retroperitoneala, submandibulära och cervikala lymfkörtlar, växer över, bildas ett angiomyomatöst hamartom i en lymfkörtel eller ett nodulärt angiomyomatöst hamartom - med partiell eller fullständig ersättning av dess parenkym. [ 25 ], [ 26 ]
Ett hamartom i huden
Vid tuberös skleros eller neurofibromatos observeras olika hudhamartom, oftast i form av hypopigmenterade fläckar; kaffe- och mjölkfläckar; angiofibrom (på kinderna, hakan, nasolabialvecken); shagreenfläckar av olika lokaliseringar (som är bindvävsnevi); fibrösa plack på pannan, hårbotten eller nacken.
En sällsynt dermatologisk manifestation av tuberös skleros (särskilt hos män) är follikulocystiskt och kollagent hamartom, som kännetecknas av riklig kollagenavsättning i dermis, koncentrisk perifollikulär fibros och keratinfyllda trattformade subkutana cystor som ses vid histopatologisk undersökning. [ 27 ]
Med hamartom bestående av melanocyter (celler som producerar pigmentet melanin) hänvisar de flesta experter även till olika melanocytiska neoplasmer, särskilt kongenitala melanocytiska nevi, vilka representerar en abnormitet i embryogenesen.
När det gäller etiologi är hamartom som består av kärlvävnad också hemangiom i huden.
Patienter med Peutz-Jeghers-Thurens syndrom har hamartom i form av fläckig pigmentering av hud och slemhinnor - lentiginosis periorificialis
Fall av linjärt papulärt ektodermalt-mesodermalt hamartom (Hamartoma moniliformis) visar ett linjärt hudfärgat papulärt utslag på huvud, nacke och övre del av bröstet.
Och ett sebocytiskt hamartom är ett hamartom i talgkörtlarna, läs mer i publikationen - talgnevus.
Hamartom i ögat
Pigmenterade hamartomatösa lesioner i iris vid neurofibromatos typ 1 och Watsons syndrom – i form av nodulära kluster av dendritiska melanocyter – definieras som irishamartom eller Lisch-noduler. De är transparenta (vanligtvis synfria), rundade, kupolformade gulbruna papler som sticker ut ovanför irisytan.
Och patienter med juvenilt angiofibrom i nasofarynx och familjär adenomatös polypos utvecklar ofta kombinerat hamartom i näthinnan och retinalt pigmentepitel – i form av en svart fläck på den centrala (makula) delen av näthinnan. [ 28 ]
Ett hamartom i näsan
Nästäppa definieras av specialister som nasalt kondromesenkymalt hamartom eller nasalt kondrom, på grund av godartad proliferation av respiratoriskt epitel, submukosala körtlar och kondrobenmesenkym. Dess kliniska manifestationer beror på lesionens storlek och lokalisering och inkluderar: nästäppa, svårigheter att andas och amma hos spädbarn, klar, vattnig nässekret och näsblödning. Ett hamartom kan växa med barnet och sprida sig till ögonhålorna, vilket resulterar i framåt- eller bakåtförskjutning av ögongloben, strabismus eller okulomotoriska störningar. [ 29 ]
Ett hamartom hos ett barn
Alla ovan nämnda hamartomatösa lesioner i olika organ och anatomiska strukturer förekommer hos barn med motsvarande syndrom.
Nyfödda uppvisar mesenkymalt hamartom i bröstväggen eller broskigt hamartom i revbenet, vilka är fasta, orörliga massor som uppstår på grund av fokal överväxt av normala skelettelement med broskiga, vaskulära och mesenkymala element. Detta hamartom kan orsaka andningssvikt och utveckling av andnödssyndrom. Mesenkymalt hamartom i levern är den näst vanligaste godartade levertumören hos barn. Denna tumörliknande formation (oftare lokaliserad i organets högra lob) består av celler från mesenkymalt stroma, hepatocyter och epitelceller i gallgångens slemhinna. Den kliniska bilden inkluderar palpabla massar i bukhålan, anorexi och viktminskning, och vid betydande storlek (upp till 10 cm och mer) täcker tumören extrahepatiska gallgångar och nedre hålvenen, vilket leder till gulsot och ödem i nedre extremiteter.
Ett hamartom är ett kongenitalt mesoblastiskt nefrom (förekommer hos 1 av 200 000 spädbarn) som kan leda till uppblåsthet i magen hos det nyfödda barnet med en palpabla massa av tät konsistens i övre högra kvadranten av buken. Spädbarn kan också uppvisa snabb ytlig andning.
Sällsynta medfödda missbildningar inkluderar fibröst hamartom i spädbarnsåldern, vilket förekommer hos barn under de första två levnadsåren och presenterar sig som en smärtfri nodulär massa i subkutana vävnader i armhålan, nacken, axeln och underarmen, ryggen och bröstet, låret, foten och de yttre könsorganen.
Ekkrin angiomatös hamartom hos ett barn kan förekomma vid födseln eller manifestera sig i tidig barndom. Denna godartade tumör av hamartomatös natur har vanligtvis utseendet som blåaktiga eller brunaktiga knölar och/eller plack som är ett resultat av proliferation av ekkrin svettkörtelvävnad och kapillärer i mellersta och djupa lagren av dermis. Detta hamartom kan orsaka lokal hyperhidros och ökad hårväxt.
Komplikationer och konsekvenser
Det är allmänt känt att hamartom sällan återkommer eller omvandlas till maligna tumörer. De uppvisar ofta få eller inga symtom och försvinner ibland till och med med tiden. Men i svårare fall och beroende på var de bildats kan dessa missbildningar få allvarliga komplikationer och konsekvenser.
Först och främst kan ett hamartom växa till en sådan storlek att det börjar trycka på omgivande vävnader och organ, vilket stör deras funktioner.
Hjärthamartom hos barn kan leda till ihållande hjärtrytmrubbningar, klaffdefekter och nedsatt intrakardiellt blodflöde med efterföljande hjärtsvikt.
Komplikationer av hamartomatösa polyper i mag-tarmkanalen är gastrointestinal blödning, obstruktion och intestinal invagination (med möjlig dödlig utgång). Och ett stort njurhamartom kan framkalla njurruptur.
Ett hamartom i hjärnan kan orsaka obstruktivt hydrocefalussyndrom.
Vid hypotalamiska och hypofysiska hamartom kan produktionen av somatotropiskt hormon (tillväxthormon) vara nedsatt, vilket kan leda till utveckling av hypofysär nanism (hypopituitarism) hos barn. Hypotalamiska hamartom hos barn kan också leda till läkemedelsresistent epilepsi.
Komplikationer av retinalt pigmentepitelhamartom är förenade med dysfunktion i näthinne- och/eller synnerven, makulaödem, neovaskularisering av åderhinnan och näthinneavlossning.
Diagnostik hamartom
En viktig del av diagnosen av hamartom och relaterade syndrom är insamling av anamnes, inklusive familjehistoria.
Laboratorietester inkluderar blodprover: allmän klinisk information; serumelektrolyter; lymfocytprofil; kalcium-, kalium-, fosfat- och ureanivåer; samt leverfunktionstester. Om möjligt utförs en finnålsaspirationsbiopsi av massan, eftersom histologisk undersökning är avgörande för diagnos och val av behandlingstaktik.
Instrumentell diagnostik ger visualisering av hamartomatös tumörliknande bildning och identifiering av dess exakta lokalisering, för vilket röntgen, angiografi, elektroencefalografi (EEG), ultraljud (sonografi), CT (datortomografi), PET (positronemissionstomografi), MRI (magnetisk resonanstomografi) används.
Differentiell diagnos
Vid alla onormala massor är differentialdiagnos mycket viktig. Således differentieras tuberkulom och hamartom; pulmonellt hamartom och primär lungcancer, bronkogen karcinoid, metastatisk sjukdom. Hjärnhamartom bör särskiljas från kraniofaryngiom och hypotalamus-kiasmatiskt gliom. Och differentialdiagnosen av hamartom som kongenitalt mesoblastiskt nefrom inkluderar Wilms tumör (malignt nefroblastom), klarcelligt sarkom i njuren och ossifierande njurtumör hos spädbarn.
Vem ska du kontakta?
Behandling hamartom
Om hamartomet är asymptomatiskt och upptäcks av en slump krävs ingen behandling, men det är nödvändigt att övervaka dess "beteende" och patientens tillstånd. I andra fall syftar behandlingen till att minska symtomintensiteten och förebygga komplikationer. Till exempel, vid hypotalamiskt hamartom med symtom på för tidig pubertet, förskrivs vissa läkemedel som hämmar frisättningen av vissa hormoner. Hjärtmediciner används för att behandla symtom på hjärtsvikt hos patienter med hjärthamartom.
Kirurgiskt avlägsnande av hamartom är indicerat för att bekräfta diagnosen och i fall av medicinskt okorrigerbara intensiva symtom.
Till exempel kan lunghamartom opereras genom kilresektion och, i svåra fall, genom att en lunglob avlägsnas (lobektomi). Ett brösthamartom kan också excideras, och om det är stort kan en partiell eller fullständig mastektomi krävas.
Stereotaktisk radiofrekvenstermoablation eller laserablation kan användas för att avlägsna hamartomatösa polyper. Radiokirurgi med högfokuserade gammastrålar – gammakniv för hypotalamiska hamartom eller astrocytiska hamartom – används också.
Förebyggande
Den enda metoden för att förhindra utvecklingen av hamartom kan betraktas som genetisk screening av barnets framtida föräldrar.
Prognos
Den övergripande prognosen för denna medfödda anomali beror på lokaliseringen och storleken av neoplasmen, såväl som på komorbiditeter och patientens allmänna hälsa.