Medicinsk expert av artikeln

Nya publikationer
Prioner - orsakande agens för prionsjukdomar
Senast recenserade: 06.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
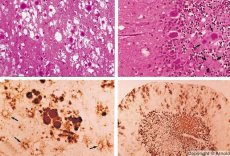
Långsamma virusinfektioner kännetecknas av speciella kriterier:
- en ovanligt lång inkubationsperiod (månader, år);
- en specifik lesion av organ och vävnader, främst det centrala nervsystemet;
- långsam, stadig progression av sjukdomen;
- oundviklig dödlig utgång.

Vissa patogener som orsakar akuta virusinfektioner kan också orsaka långsamma virusinfektioner. Till exempel orsakar mässlingsvirus ibland SSPE, och röda hund-virus orsakar progressiv kongenital röda hund och röda hund-panencefalit.
En typisk långsam virusinfektion hos djur orsakas av visna/madi-viruset, som är ett retrovirus. Det är orsaken till långsam virusinfektion och progressiv lunginflammation hos får. Hjärnans vita substans förstörs, förlamning utvecklas (visna - förtvining); kronisk inflammation i lungor och mjälte uppstår.
Sjukdomar som i sina egenskaper liknar långsamma virusinfektioner orsakas av prioner - de orsakande agenserna för prioninfektioner. Prionsjukdomar är en grupp progressiva sjukdomar i centrala nervsystemet hos människor och djur. Hos människor försämras centrala nervsystemets funktion, personlighetsförändringar sker och rörelsestörningar uppstår. Sjukdomens symtom varar vanligtvis från flera månader till flera år och slutar med döden. Tidigare betraktades prioninfektioner tillsammans med de så kallade orsakande agenserna för långsamma virusinfektioner.
Vissa agens som orsakar prionsjukdomar ackumuleras först i lymfoida vävnader. Prioner, som kommer in i hjärnan, ackumuleras i stora mängder, vilket orsakar amyloidos (extracellulär dysproteinos, kännetecknad av amyloidavsättning med utveckling av atrofi och skleros i vävnaden) och astrocytos (proliferation av astrocytiska neuroglia, hyperproduktion av gliafibrer). Fibriller, aggregat av protein eller amyloid och spongiforma förändringar i hjärnan (överförbara spongiforma encefalopatier) bildas. Som ett resultat förändras beteendet, koordinationen av rörelser försämras, utmattning med dödlig utgång utvecklas. Immunitet bildas inte. Prionsjukdomar är konformationssjukdomar som utvecklas till följd av felaktig vikning (brott i korrekt konformation) av cellulärt protein som är nödvändigt för kroppens normala funktion. Prionöverföringsvägarna varierar:
- matsmältningsväg - infekterade produkter av animaliskt ursprung, livsmedelstillsatser från råa nötkreatursorgan etc.:
- överföring genom blodtransfusion, administrering av läkemedel av animaliskt ursprung, organ- och vävnadstransplantation, användning av infekterade kirurgiska och dentala instrument;
- överföring genom immunobiologiska preparat (infektion av 1500 får med PrP''' med hjärnformolvaccin från sjuka får är känd).
Patologiska prioner, som har kommit in i tarmen, transporteras till blodet och lymfan. Efter perifer replikering i mjälten, blindtarmen, tonsillerna och andra lymfoida vävnader överförs de till hjärnan genom perifera nerver (neuroinvasion). Direkt penetration av prioner in i hjärnan genom blod-hjärnbarriären är möjlig. Tidigare trodde man att det centrala nervsystemet var den enda vävnaden där patologiska prioner ackumuleras, men studier har dykt upp som har ändrat denna hypotes. Det visade sig att ackumuleringen av prioner i mjälten är förknippad med ökningen och funktionen av follikulära dendritiska celler.
 [
[ Egenskaper hos prioner
Den normala cellulära isoformen av prionproteinet med en molekylvikt på 33-35 kDa bestäms av prionproteingenen (priongenen - PrNP är belägen på den 20:e mänskliga kromosomen). Den normala genen förekommer på cellytan (förankrad i membranet av molekylens glykoprotein), känslig för proteas. Den reglerar överföringen av nervimpulser, dagliga cykler, oxidationsprocesser, deltar i kopparmetabolismen i centrala nervsystemet och i regleringen av benmärgsstamcellsdelning. Dessutom finns priongenen i mjälten, lymfkörtlar, hud, mag-tarmkanal och follikulära dendritiska celler.
Spridning av patologiska prioner
Omvandlingen av prioner till förändrade former sker när den kinetiskt kontrollerade jämvikten mellan dem störs. Processen förstärks av en ökning av mängden patologiska (PrP) eller exogena prioner. PrP är ett normalt protein förankrat i cellmembranet. PrP' är ett globulärt hydrofobt protein som bildar aggregat med sig självt och PrP'' på cellytan: som ett resultat omvandlas PrP' till PrP'' och sedan fortsätter cykeln. Den patologiska formen av PrP''' ackumuleras i neuroner, vilket ger cellen ett svampigt utseende.
Kuru
Prionsjukdom, tidigare vanlig bland papuanerna (som betyder darrning eller skakningar) i den östra delen av ön Nya Guinea. Sjukdomens smittsamma egenskaper bevisades av K. Gajdusek. Patogenen överförs via mat som ett resultat av rituell kannibalism - att äta den otillräckligt tillagade, prioninfekterade hjärnan hos avlidna släktingar. Som ett resultat av skador på det centrala nervsystemet försämras rörelse och gång, frossa och eufori ("skrattdöd") uppstår. Inkubationsperioden varar 5-30 år. Patienten dör efter ett år.
Creutzfeldt-Jakobs sjukdom
Prionsjukdom, som manifesterar sig som demens, syn- och lillhjärnsrubbningar samt rörelserubbningar med dödlig utgång efter 4–5 månaders sjukdom vid den klassiska varianten av Creutzfeldt-Jakobs sjukdom och efter (3–14 månader vid den nya varianten av Creutzfeldt-Jakobs sjukdom). Inkubationstiden kan uppgå till 20 år. Olika smittvägar och orsaker till sjukdomen är möjliga:
- vid konsumtion av otillräckligt värmebehandlade animaliska produkter, såsom kött och hjärnor från kor med bovin spongiform encefalopati;
- vid vävnadstransplantation, såsom hornhinnetransplantation, blodtransfusion, användning av hormoner och andra biologiskt aktiva substanser av animaliskt ursprung, användning av katgut, kontaminerade eller otillräckligt steriliserade kirurgiska instrument, manipulationer av prosektorer;
- vid hyperproduktion av PrR och andra tillstånd som stimulerar processen att omvandla PrR' till PrR".
Sjukdomen kan också utvecklas som ett resultat av en mutation eller insättning i prion-genregionen. Sjukdomen är vanlig i familjär karaktär på grund av genetisk predisposition för Creutzfeldt-Jakobs sjukdom. I den nya varianten av Creutzfeldt-Jakobs sjukdom utvecklas sjukdomarna vid en yngre ålder (medelålder 28 år), till skillnad från den klassiska varianten (medelålder 65 år). I den nya varianten av Creutzfeldt-Jakobs sjukdom ackumuleras onormalt prionprotein inte bara i centrala nervsystemet utan även i lymforetikulära vävnader, inklusive tonsillerna.
Gerstmann-Sträussler-Scheinkers syndrom
Ärftlig prionsjukdom, åtföljd av demens, hypotoni, sväljningssvårigheter (dysfagi), dysartri. Har ofta en familjär karaktär. Inkubationstiden är från 5 till 30 år. Sjukdomen uppträder vid 50-60 års ålder, dess varaktighet varierar från 5 till 13 år.
Ärftlig dödlig sömnlöshet
En autoimmun sjukdom med progressiv sömnlöshet, sympatisk hyperreaktivitet (hypertoni, hypertermi, hyperhidros, takykardi), tremor, ataxi, multiklon, hallucinationer. Sömnen är allvarligt störd. Döden inträffar med progression av kardiovaskulär svikt.
Skrapa
Scrapie (från engelskans scrape - att skrapa) är en prionsjukdom hos får och getter (skabb), som uppstår med skador på centrala nervsystemet, progressiva rörelsestörningar, svår klåda i huden (skabb) och slutar med djurets död.
Bovin spongiform encefalopati
En sjukdom hos nötkreatur som kännetecknas av skador på centrala nervsystemet, försämrad koordination av rörelser och oundviklig död hos djuret. Sjukdomens epidemi utbröt först i Storbritannien. Den var förknippad med att djuren utfodrades med kött- och benmjöl innehållande patologiska prioner. Inkubationstiden varierar från 1,5 till 15 år. Hjärnan, ryggmärgen och ögonen hos djuren är mest infekterade.
Laboratoriediagnostik av prionsjukdomar
Under diagnostiken noteras spongiforma förändringar i hjärnan, astrocytos (glios) och avsaknad av inflammatoriska infiltrat. Hjärnan färgas för amyloid. Proteinmarkörer för prionsjukdomar i hjärnan detekteras i cerebrospinalvätskan (med ELISA). Genetisk analys av priongenen (PCR) utförs.
Förebyggande av prionsjukdomar
Autoklavering (vid 134 °C i 18 minuter; vid 121 °C i 1 timme), förbränning, ytterligare behandling med blekmedel och en 1-N NaCl-lösning i 1 timme rekommenderas för dekontaminering av instrument och föremål i omgivningen. För ospecifik profylax har restriktioner införts för användning av läkemedel av animaliskt ursprung och produktion av hypofyshormoner av animaliskt ursprung är förbjuden. Transplantation av dura mater är begränsad. Gummihandskar används vid arbete med patienters dialogiska vätskor.