Medicinsk expert av artikeln

Nya publikationer
Neuroblastom hos barn: orsaker, diagnos, behandling
Senast recenserade: 04.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
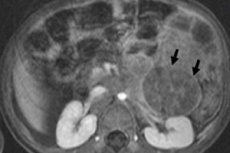
Inom pediatrisk onkologi är en av de vanligaste extrakraniella neoplasmerna neuroblastom hos barn, vilket är en embryonal malign tumör i neurala crest-neuroblaster, det vill säga embryonala (omogna) nervceller i det sympatiska nervsystemet.
Epidemiologi
Enligt statistik från International Neuroblastoma Risk Group (INRG) står neuroblastom för cirka 8 % av alla onkologiska sjukdomar hos barn världen över och är den tredje vanligaste förekomsten, efter leukemi och hjärntumörer.
Enligt andra uppgifter står neuroblastom för cirka 28 % av alla cancerfall hos spädbarn. Mer än en tredjedel av fallen av neuroblastom diagnostiseras hos barn under ett år; den genomsnittliga diagnosåldern är 19–22 månader. Mer än 90 % av de diagnostiserade fallen förekommer hos barn i åldern två till fem år (med en övervikt av pojkar); den högsta incidensen observeras vid två till tre års ålder, och fall hos barn över fem år står för mindre än 10 %.
Orsaker neuroblastom
I studier av orsakerna till neuroblastom har forskare dragit slutsatsen att denna tumör hos barn uppstår på grund av sporadiska genetiska mutationer under embryogenesen eller tidig postnatal utveckling. Men orsakerna till dessa genförändringar är okända, eftersom ingen påverkan av teratogena miljöfaktorer har identifierats.
Dessa tumörer kan förekomma var som helst, inklusive mediastinum, nacke, buk, binjurar, njurar, ryggrad och bäcken.
I sällsynta fall kan neuroblastom hos spädbarn vara associerat med en ärftlig mutation. I synnerhet en mutation i genen för membranproteinet CD246 på kromosom 2 - enzymet tyrosinkinas ALK, vilket säkerställer intercellulär kommunikation och spelar en viktig roll i nervsystemets funktion; i genen för proteinet PHOX2B (på kromosom 4), vilket är involverat i mognaden av nervceller.
Neuroblastom kan också vara associerat med neurofibromatos typ 1 i barndomen,Beckwith-Wiedemanns syndrom och hyperinsulinemisk hypoglykemi (nesidioblastos i pankreatit).
Riskfaktorer
Idag erkänns ärftlighet som riskfaktorer för utveckling av neuroblastom hos barn - förekomsten av denna tumör i familjehistorien, såväl som medfödda missbildningar i samband med genmutationer under intrauterin utveckling. Detta gäller särskilt för fall av utveckling av flera neoplasmer i olika organ.
Inga av de exogena faktorer som ökar risken för denna tumör har identifierats av forskare.
Patogenes
Utvecklingsmekanismen för neuroblastom orsakas av störningar i differentiering och mognad av neural crest-celler – bilaterala cellinjer som bildas vid kanterna av neuralröret från det ektodermala groddskiktet hos det mänskliga embryot. Dessa celler migrerar (förflyttar sig) och differentierar till många typer av celler: sensoriska och autonoma neuroner, neuroendokrina celler och celler i binjuremärgen, celler i kraniofacialt brosk och ben, samt pigmentceller.
Vid neuroblastom mognar inte de migrerade neuroblasterna, utan fortsätter att växa och dela sig och bilda en tumör. Och patogenesen för dess bildning är förknippad med följande genmutationer:
- med duplicering av en del av kromosomsekvensen eller duplicering av segment av LMO1-genen på kromosom 11, som kodar för RBTN1-proteinet i embryots neurala crestceller;
- med en förändring i kopietalet av NBPF10-genen på kromosom 1q21.1, som kodar för DUF1220-proteinet, vilket kontrollerar proliferationen av mänskliga neurala stamceller. Dessa störningar leder antingen till en duplicering av denna kromosom eller till dess deletion - frånvaron av en del av DNA:t;
- med förändringar i tumörsuppressorgenen ATRX (på kromosom Xq21.1);
- med närvaron av ytterligare kopior (amplifiering) av N-Myc-transkriptionsfaktorgenen på kromosom 2, som kodar för en av transkriptionsfaktorerna (DNA-bindande protein) som reglerar aktiviteten hos andra gener och kontrollerar proliferationen av prekursorceller under bildandet av proteiner för bildandet av fostrets vävnader och organ. Amplifiering av denna gen omvandlar den till en onkogen, vilket provocerar en störning av cellcykeln, ökad cellproliferation och tumörbildning.
Symtom neuroblastom
De första tecknen på neuroblastom är ospecifika och kan inkludera aptitlöshet (och viktminskning), trötthet vid matning, feber och ledvärk.
Kliniska symtom beror på den primära tumörens lokalisation och förekomsten av metastaser (som förekommer i 60-73% av fallen).
Mycket ofta är primärt neuroblastom lokaliserat i binjuremärgen, som har ett liknande ursprung som nervceller. Hos barn under ett år diagnostiseras binjurebarksneuroblastom i 35–40 % av fallen. Symtomen inkluderar buksmärtor, feber, viktminskning, skelettsmärta, anemi eller samtidig Peppers syndrom: diffus leverskada med svår hepatomegali och andnödssyndrom.
Retroperitonealt neuroblastom eller retroperitonealt neuroblastom hos barn börjar, allt eftersom det växer, trycka på urinblåsan eller tarmarna, vilket kan orsaka problem med urinering eller avföring, svullnad i benen (hos pojkar svullnar pungen).
Neuroblastom i mediastinum hos barn (mediastinalt neuroblastom) trycker ofta på vena cava superior, vilket kan orsaka svullnad i ansikte, hals, armar och övre delen av bröstet (där huden blir blåröd med subkutana knutor). Hosta och väsande andning, andningssvårigheter (andnöd) eller sväljningssvårigheter (dysfagi) uppträder; förstorade lymfkörtlar observeras på halsen, ovanför nyckelbenet och i armhålorna.
Spridningen av tumörceller till benmärgen leder till anemi, trombocytopeni och leukopeni med blödningstendens.
Och med metastaser i det periorbitala området uppstår mörka ringar eller blåmärken runt ögonen. En sådan tumör kan också orsaka huvudvärk och yrsel, exoftalmi (utbuktning av ögongloberna), och på grund av kompression av nervändarna - hängande ögonlock (ptos) och en minskning av pupillstorleken (mios).
Abdominellt neuroblastom eller neuroblastom i bukhålan hos barn leder till bildandet av palpabla tätningar i buken, dess utspändhet, aptitlöshet, förstoppning och förhöjt blodtryck. En tumör som trycker på ryggmärgen eller nervroten kan leda till domningar och svaghet i armar och ben, oförmåga att stå, krypa eller gå. Om benen påverkas kan skelettsmärta uppstå.
Vid tumör i stadium 3-4 i bukhålan med lymfkörtelskador kan tumörceller komma in i njurparenkymet, och då utvecklas omfattande neuroblastom i njuren hos barn, vilket leder till störningar i dess funktioner.
Stages
- Steg 1 neuroblastom är en primär tumör som är lokaliserad och isolerad till ett område av kroppen; lymfkörtlar på båda sidor påverkas inte.
- Neuroblastom stadium 2. I stadium 2A är den primära tumören begränsad till ett område men är stor; bilaterala lymfkörtlar är inte involverade. I stadium 2B är lymfkörtlarna på den sida av kroppen där tumören är belägen positiva för metastaser.
- Neuroblastom stadium 3: den primära tumören korsar ryggmärgen eller kroppens mittlinje, unilaterala eller bilaterala metastaser finns i lymfkörtlarna.
- Neuroblastom stadium 4: tumören har spridit sig till avlägsna lymfkörtlar, benmärg, skelett, lever eller andra organ. Och stadium 4S diagnostiseras hos barn under ett år med en lokaliserad primärtumör, med spridning till hud, lever eller benmärg.
Internationella systemet för riskstadieindelning av neuroblastom (INRGSS)
INRGSS använder bilddefinierade riskfaktorer (IDRF), vilka är faktorer som ses vid bilddiagnostiska undersökningar och som kan innebära att en tumör blir svårare att avlägsna.
INRGSS delar in neuroblastom i fyra stadier:
- L1: Tumören har inte spridit sig från där den började och har inte vuxit till vitala strukturer. Den är begränsad till en del av kroppen, såsom halsen, bröstet eller buken.
- L2: Tumören har inte spridit sig (metastaserat) långt från där den började (till exempel kan den ha vuxit från vänster sida av buken till vänster sida av bröstet), men den har minst en IDRF.
- M: Tumören har metastaserat till en avlägsen del av kroppen (förutom tumörer i MS-stadiet).
- MS: Metastatisk sjukdom hos barn under 18 månader, där cancern endast har spridit sig till huden, levern och/eller benmärgen.
Komplikationer och konsekvenser
Neuroblastom kännetecknas av komplikationer och konsekvenser såsom:
- spridning (metastasering) till lymfkörtlar, benmärg, lever, hud och ben;
- kompression av ryggmärgen (vilket kan orsaka smärta och leda till förlamning);
- utveckling av paraneoplastiskt syndrom (på grund av verkan av vissa kemikalier som utsöndras av tumören, såväl som antigenet disialogangliosid GD2 som uttrycks av dess celler), vilket manifesteras av snabba ofrivilliga ögonrörelser, nedsatt koordination, muskelkramper och diarré;
- återfall efter avslutad primärbehandling (som klinisk praxis visar har högriskneuroblastom ett återfall i 50 % av fallen).
Diagnostik neuroblastom
Diagnos av misstänkt neuroblastom hos ett barn kräver undersökning, laboratorietester och bilddiagnostik.
Blod- och urinprov tas för katekolaminer (norepinefrin och dopamin) och homovanill- eller vanillylmandelsyror (som bildas under metabolismen av dessa hormoner); ett blodprov för neurospecifik enolas, en enzymlänkad immunosorbentanalys (ELISA) av blodserum och en benmärgsanalys (varav ett prov tas genom aspirationspunktion). Ett DNA-test utförs för att fastställa mutationer och en biopsi utförs för cytomorfologisk undersökning av tumörvävnaden.
Efter att biopsiproverna har tagits skickas de till ett laboratorium där de undersöks i mikroskop av en patolog (en läkare som har specialutbildning i att identifiera cancerceller). Speciella laboratorietester utförs ofta också på proverna för att visa om tumören är neuroblastom.
Om det är neuroblastom kan laboratorietester också hjälpa till att avgöra hur snabbt tumören kan växa eller sprida sig, samt vilka behandlingar som kan fungera bäst.
Instrumentell diagnostik visualiserar tumören med hjälp av ultraljud, röntgen, MRI eller CT, PET med introduktion av 18F-fluorodeoxyglukos eller MIBG-skanning - scintigrafi med metajodbensylguanidin. [ 1 ]
Differentiell diagnos
Differentialdiagnos inkluderar benignt ganglioneurom, ganglioneuroblastom, rabdomyosarkom och nefroblastom.
Vem ska du kontakta?
Behandling neuroblastom
Vid neuroblastom beror behandlingen på patientens riskgrupp (tumörprocessens stadium), tumörens lokalisering, tumörcellernas genomiska egenskaper och barnets ålder. Och den kan inkludera övervakning, kirurgi, kemoterapi, strålbehandling, immunterapi och hematopoetisk stamcellstransplantation.
Neoadjuvant eller adjuvant (pre- eller postoperativ) kemoterapi för neuroblastom hos barn, liksom all kemoterapi för cancer, ges i kurer: läkemedlet administreras under flera dagar i rad, följt av ett uppehåll för att kroppen ska kunna återhämta sig. Cykler upprepas vanligtvis var tredje till fjärde vecka.
Följande läkemedel (och deras kombinationer) används: cyklofosfamid, cisplatin eller karboplatin, doxorubicin (adriamycin), vinkristin, etoposid.
Vanliga biverkningar av cellgiftsbehandling inkluderar håravfall, aptitlöshet, trötthet, illamående och kräkningar, munsår, diarré eller förstoppning. Cellgiftsbehandling kan skada benmärgen och orsaka en minskning av antalet blodkroppar.
Riktad immunterapi (riktad mot tumörantigenet GD2) använder läkemedel från gruppen monoklonala antikroppar (anti-GD2 MAb) Dinutuximab (Unituxin) och Naxitamab. De administreras intravenöst genom långvarig infusion, i kombination med granulocyt-makrofagkolonistimulerande faktor (cytokin GM-CSF) och interleukin-2.
Biverkningar av dessa läkemedel inkluderar smärta (ofta mycket svår), sänkt blodtryck, ökad hjärtfrekvens, andnöd (med eventuell svullnad i luftvägarna), ökad temperatur, illamående, kräkningar och diarré, förändringar i blodets cell- och mineralsammansättning.
För att minska risken för återfall av cancer efter högdoserad kemoterapi och stamcellstransplantation behandlas barn med högriskneuroblastom med systemiska retinoider, 13-cis-retinsyra (isotretinoin). [ 2 ]
Kirurgisk behandling av neuroblastom – tumörborttagning, till exempel öppen adrenalektomi eller laparoskopisk resektion av binjurebarksneuroblastom; lymfektomi (borttagning av drabbade lymfkörtlar) etc. [ 3 ]
Vid högriskneuroblastom kan strålbehandling användas.[ 4 ]
Förebyggande
Med tanke på orsakerna till neuroblastom hos barn kan den enda förebyggande åtgärden vara genetisk rådgivning vid graviditetsplanering. Men man bör komma ihåg att denna tumör endast är associerad med ärftliga mutationer i 1-2% av fallen.
Prognos
Infantilt neuroblastom har förmågan att spontant regressera.
Prognostiska markörer
- Högrisktumörer, liksom neuroblastom hos barn i alla åldersgrupper och alla stadier (förutom stadium 4S) – med ökat uttryck av N-MYC-genen och amplifiering av N-Myc-onkogenen – har en ogynnsam prognos som påverkar den förväntade livslängden.
- Att ha tumörceller som saknar vissa delar av kromosom 1 eller 11 (kända som 1p- eller 11q-deletioner) har en sämre prognos. Att ha en extra del av kromosom 17 (17q-förstärkning) är också förknippat med en sämre prognos.
- Neuroblastomceller med en stor mängd DNA har en bättre prognos, särskilt för barn under 2 år.
- Neuroblastom som har fler neurotrofinreceptorer, särskilt nervtillväxtfaktorreceptorn TrkA, har en bättre prognos.
Överlevnad per riskgrupp för barndomsonkologi (COG)
- Lågriskgrupp: Barn i lågriskgruppen har en 5-årsöverlevnad på över 95 %.
- Mellanriskgrupp: Barn i mellanriskgruppen har en 5-årsöverlevnad på 90 % till 95 %.
- Högriskgrupp: Barn i högriskgruppen har en 5-årsöverlevnad på cirka 50 %.
Cirka 15 % av alla dödsfall i barndomen i samband med cancer beror på neuroblastom. Chanserna till långsiktig överlevnad för denna högriskmalignitet är högst 40 %. Den totala femårsöverlevnaden är 67–74 %, 43 % i åldersgruppen ett till fyra år och mer än 80 % för neuroblastom som diagnostiseras under det första levnadsåret.
Использованная литература