Medicinsk expert av artikeln

Nya publikationer
Charcot-Marie-Tooths sjukdom.
Senast recenserade: 12.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
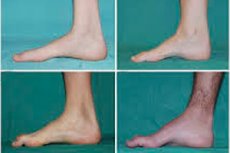
Peroneal muskelatrofi, Charcot-Marie-Tooths syndrom eller sjukdom är en grupp kroniska ärftliga sjukdomar med skador på perifera nerver.
Enligt ICD-10, i avsnittet om sjukdomar i nervsystemet, är koden för denna sjukdom G60.0 (ärftlig motorisk och sensorisk neuropati). Den ingår också i listan över sällsynta sjukdomar.
Epidemiologi
Enligt klinisk statistik är förekomsten av alla typer av Charcot-Marie-Tooth-sjukdom per 100 tusen av befolkningen 19 fall (enligt andra källor, ett fall per 2,5-10 tusen av befolkningen).
CMT typ 1 står för ungefär två tredjedelar av fallen (ett fall per 5 000 till 7 000 invånare), och nästan 70 % av dem är associerade med duplicering av PMP22-genen. Mer än 1,2 miljoner människor världen över lider av denna typ av sjukdom.
Incidensen av CMT typ 4 uppskattas till 1–5 fall per 10 000 barn. [ 1 ]
Orsaker Charcot-Marie-Tooths sjukdom
Enligt klassificeringen av polyneuropatiska syndrom avser peroneal (fibulär) muskelatrofi, Charcot-Marie-Tooths neurala amyotrofi eller Charcot-Marie-Tooths sjukdom (förkortat CMT) genetiskt betingade motorisk-sensoriska polyneuropatier. [ 2 ]
Det vill säga, orsakerna till dess uppkomst är genetiska mutationer. Och beroende på arten av genetiska avvikelser skiljer man sig mellan huvudtyperna eller typerna av detta syndrom: demyeliniserande och axonala. Den första gruppen inkluderar Charcot-Marie-Tooths sjukdom typ 1 (CMT1), som uppstår på grund av duplicering av PMP22-genen på kromosom 17, som kodar för transmembran perifert myelinprotein 22. Som ett resultat sker segmentell demyelinisering av axonmanteln (nervcellsprocesser) och en minskning av hastigheten på nervsignalledning. Dessutom kan mutationer finnas i vissa andra gener.
Den axonala formen är Charcot-Marie-Tooths sjukdom typ 2 (CMT2), som drabbar själva axonerna och är associerad med patologiska förändringar i MFN2-genen vid lokus 1p36.22, som kodar för membranproteinet mitofusin-2, vilket är nödvändigt för mitokondriefusion och bildandet av funktionella mitokondriella nätverk inuti perifera nervceller. Det finns mer än ett dussin subtyper av CMT2 (med mutationer i specifika gener).
Det bör noteras att för närvarande har mer än hundra gener identifierats vars skador, som överförs genom ärftlighet, orsakar olika subtyper av Charcot-Marie-Tooths sjukdom. Till exempel leder mutationer i RAB7-genen till CMT typ 2B; förändring av SH3TC2-genen (som kodar för ett av membranproteinerna i Schwann-celler) orsakar CMT typ 4C, som manifesterar sig i barndomen och kännetecknas av demyelinisering av motoriska och sensoriska neuroner (det finns ett dussin och ett halvt former av typ 4 av denna sjukdom).
En sällsynt typ 3 CMT (kallad Dejerine-Sottas syndrom) börjar utvecklas i tidig barndom och orsakas av mutationer i PMP22, MPZ, EGR2 och andra gener.
När CMT typ 5 uppstår vid 5-12 års ålder observeras inte bara motorisk neuropati (i form av spastisk parapares i nedre extremiteterna), utan även skador på syn- och hörselnerverna.
Muskelsvaghet och optisk atrofi (med synförlust) samt balansproblem är typiska för CMT typ 6. Och vid typ 7 Charcot-Marie-Tooths sjukdom finns inte bara motorisk-sensorisk neuropati, utan även en näthinnesjukdom i form av retinitis pigmentosa.
X-länkad CMT eller Charcot-Marie-Tooths sjukdom med tetrapares i extremiteterna (rörlighetssvaghet i både armar och ben) är vanligare bland män och är en demyeliniserande typ och tros bero på en mutation i GJB1-genen på den långa armen av X-kromosomen, som kodar för connexin 32, ett transmembranprotein i Schwann-celler och oligodendrocyter som reglerar överföringen av nervsignaler. [ 3 ]
Riskfaktorer
Den huvudsakliga riskfaktorn för CMT är en familjehistoria av sjukdomen, det vill säga hos nära släktingar.
Enligt genetiker är risken att få ett barn som utvecklar sjukdomen 25 % om båda föräldrarna är bärare av den autosomalt recessiva genen för Charcot-Marie-Tooths sjukdom. Och risken att barnet är bärare av denna gen (men inte har några symtom) uppskattas till 50 %.
Vid X-länkad nedärvning (när den muterade genen finns på kvinnans X-kromosom) finns det 50 % risk att modern för genen vidare till sin son, som kommer att utveckla CMT. Sjukdomen kanske inte uppstår när ett flickebarn föds, men dotterns söner (barnbarn) kan ärva den defekta genen, och sjukdomen kommer att utvecklas.
Patogenes
Vid alla typer av Charcot-Marie-Tooth-sjukdom orsakas dess patogenes av en ärftlig anomali i perifera nerver: motoriska (rörelse) och sensoriska (känsliga).
Om CMT-typen är demyeliniserande, leder förstörelsen eller defekten av myelinskidan som skyddar axonerna i perifera nerver till en avmattning i överföringen av nervimpulser i det perifera nervsystemet - mellan hjärnan, musklerna och sensoriska organ.
Vid den axonala typen av sjukdomen påverkas själva axonerna, vilket negativt påverkar nervsignalernas styrka, vilket är otillräckligt för fullständig stimulering av muskler och sensoriska organ.
Läs också:
Hur överförs Charcot-Marie-Tooth syndrom? De defekta generna kan ärvas autosomalt dominant eller autosomalt recessivt.
Den vanligaste typen, autosomalt dominant nedärvning, inträffar när det finns en kopia av den muterade genen (som bärs av en av föräldrarna). Och sannolikheten att överföra CMT till varje födt barn uppskattas till 50 %. [ 4 ]
Vid autosomalt recessiv nedärvning behövs två kopior av den defekta genen (en från varje förälder som inte visar några tecken på sjukdomen) för att sjukdomen ska utvecklas.
I 40–50 % av fallen förekommer autosomalt dominant ärftlig demyelinisering, dvs. CMT typ 1; i 12–26 % av fallen axonal CMT, dvs. typ 2. Och i 10–15 % av fallen observeras X-länkad nedärvning. [ 5 ]
Symtom Charcot-Marie-Tooths sjukdom
Vanligtvis börjar de första tecknen på denna sjukdom uppstå i barndomen och tonåren och utvecklas gradvis under hela livet, även om syndromet kan göra sig känt senare. Kombinationen av symtom varierar, och sjukdomens progressionshastighet, liksom dess svårighetsgrad, är omöjlig att förutsäga.
Typiska symtom på det initiala stadiet inkluderar ökad allmän trötthet; minskad tonus (svaghet) i musklerna i fötter, vrister och smalben; brist på reflexer. Detta försvårar fotrörelser och leder till dysbasi (gångstörning) i form av högre benställning, ofta med täta snubblingar och fall. Tecken på Charcot-Marie-Tooths sjukdom hos ett litet barn kan inkludera uttalad klumpighet och åldersotypiska gångsvårigheter i samband med bilateral droppfot. Fotdeformiteter är också karakteristiska: hög fotvalv (ihålig fot) eller svåra platta fötter, böjda (hammarformade) tår.
Vid gång på tårna mot bakgrund av muskelhypotoni kan en neurolog misstänka att barnet har typ 4 CMT, där barn kanske inte kan gå i tonåren.
Allt eftersom sjukdomen fortskrider sprider sig muskelatrofi och svaghet till de övre extremiteterna, vilket gör det svårt att utföra finmotorik och normala handuppgifter. Minskade taktila förnimmelser och förmågan att känna värme och kyla, samt domningar i fötter och händer, tyder på skador på axonerna i sensoriska nerver.
Vid Charcot-Marie-Tooths sjukdom typ 3 och 6, som manifesterar sig i barndomen, observeras sensorisk ataxi (försämrad koordination av rörelser och balans), muskelryckningar och tremor, skador på ansiktsnerven, synnervsatrofi med nystagmus och hörselnedsättning.
I senare stadier kan det förekomma okontrollerbara skakningar (tremor) och frekventa muskelkramper; problem med rörelser kan leda till utveckling av smärta: muskel-, led-, neuropatisk.
Komplikationer och konsekvenser
Charcot-Marie-Tooths sjukdom kan ha komplikationer och konsekvenser såsom:
- mer frekventa stukningar och frakturer;
- kontrakturer i samband med förkortning av periartikulära muskler och senor;
- skolios (ryggradens krökning);
- andningsproblem – när nervfibrerna som innerverar diafragmans muskler är skadade:
- förlust av förmågan att röra sig självständigt.
Diagnostik Charcot-Marie-Tooths sjukdom
Diagnosen omfattar klinisk undersökning, anamnes (inklusive familjehistoria), neurologisk och systemisk undersökning.
Tester utförs för att kontrollera rörelseomfång, känslighet och senreflexer. Nervledning kan bedömas med hjälp av instrumentell diagnostik – elektromyografi eller elektroneuromyografi. Ultraljud eller MR kan också krävas. [ 6 ]
Genetiska eller DNA-tester för att upptäcka de vanligaste genetiska mutationerna som orsakar CMT i ett blodprov är begränsade eftersom DNA-tester för närvarande inte är tillgängliga för alla typer av sjukdomen. För mer information, se Genetiska tester.
I vissa fall utförs en biopsi av den perifera nerven (vanligtvis den surala nerven).
Differentiell diagnos
Differentialdiagnos inkluderar andra perifera neuropatier, Duchennes muskeldystrofi, myelopatiska och myasteniska syndrom, diabetisk neuropati, myelopatier vid multipel och amyotrofisk lateralskleros, Guillain-Barrés syndrom, trauma på peronealnerven och dess atrofi (inklusive vid klämning mellan ländryggsdiskarna), skador på lillhjärnan eller talamus, samt biverkningar av kemoterapi (under behandling med cytostatika såsom vinkristin eller paklitaxel). [ 7 ]
Vem ska du kontakta?
Behandling Charcot-Marie-Tooths sjukdom
Idag omfattar behandling av denna ärftliga sjukdom träningsterapi (som syftar till att stärka och stretcha muskler); arbetsterapi (som hjälper patienter med muskelsvaghet i händerna); och användning av ortopediska hjälpmedel för att underlätta gång. Vid behov förskrivs smärtstillande eller antikonvulsiva medel. [ 8 ]
Vid svåra platta fötter kan osteotomi utföras, och vid häldeformation är kirurgisk korrigering indicerad – artrodes. [ 9 ]
Forskning pågår både kring sjukdomens genetiska komponent och dess behandlingsmetoder. Användningen av stamceller, vissa hormoner, lecitin eller askorbinsyra har ännu inte gett positiva resultat.
Men tack vare den senaste forskningen kan något nytt verkligen dyka upp i behandlingen av Charcot-Marie-Tooths sjukdom inom en snar framtid. Således har det franska företaget Pharnext sedan 2014 utvecklat, och sedan mitten av 2019 har kliniska prövningar pågått för läkemedlet PXT3003 för behandling av CMT typ 1 hos vuxna, vilket hämmar ökat uttryck av PMP22-genen, förbättrar myelinisering av perifera nerver och lindrar neuromuskulära symtom.
Specialister från läkemedelsföretaget Sarepta Therapeutics (USA) arbetar med att skapa en genterapi för Charcot-Marie-Tooths sjukdom typ 1. Denna terapi kommer att använda ett ofarligt adenoassocierat virus (AAV) av släktet Dependovirus med ett linjärt enkelsträngat DNA-genom, vilket kommer att överföra NTF3-genen till kroppen, som kodar för neurotrofin-3 (NT-3)-proteinet som är nödvändigt för Schwann-nervcellernas funktion.
Helixmith kommer att inleda kliniska prövningar av den sydkoreanskt utvecklade genterapin Engensis (VM202) i slutet av 2020 för att behandla muskelsymtom vid CMT typ 1. [ 10 ]
Förebyggande
Förebyggande av CMT kan vara genetisk rådgivning av blivande föräldrar, särskilt om någon i paret har en familjehistoria av sjukdomen. Emellertid har fall av de novo punktgenmutationer identifierats, det vill säga i frånvaro av sjukdomen i familjehistorien.
Under graviditeten kan sannolikheten för Charcot-Marie-Tooths sjukdom hos det framtida barnet kontrolleras genom korionisk villusbiopsi (från 10 till 13 veckors graviditet), samt en analys av fostervätska (vid 15-18 veckor).
Prognos
Generellt sett beror prognosen för olika typer av Charcot-Marie-Tooths sjukdom på den kliniska svårighetsgraden, men i samtliga fall fortskrider sjukdomen långsamt. Många patienter har funktionsnedsättningar, även om detta inte minskar livslängden.