Medicinsk expert av artikeln

Nya publikationer
Cystisk fibros
Senast recenserade: 04.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
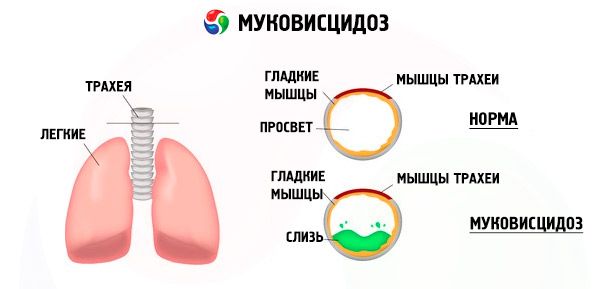
Cystisk fibros är en genetisk autosomal recessiv monogen sjukdom som kännetecknas av en störning i utsöndringen av exokrina körtlar i vitala organ med skador främst på andnings- och matsmältningssystemen, svårt förlopp och ogynnsam prognos.
 [ 1 ]
[ 1 ]
Epidemiologi
Incidensen av cystisk fibros varierar mellan 1:2 500 och 1:4 600 nyfödda. Varje år föds cirka 45 000 personer med cystisk fibros världen över. Incidensen av bärare av genen för cystisk fibros är 3–4 %, varav cirka 275 miljoner människor världen över är bärare av denna gen, varav cirka 5 miljoner bor i Ryssland och cirka 12,5 miljoner i OSS-länderna.
Orsaker cystisk fibros
Cystisk fibros överförs autosomalt recessivt. Genen för cystisk fibros finns i autosom 7, innehåller 27 exoner och består av 250 000 nukleotidpar.
En enda gen kan ha många mutationer, där var och en är specifik för en viss population eller geografisk region. Mer än 520 mutationer har beskrivits, varav den vanligaste är delta-P-508, dvs. en substitution av aminosyran fenylalanin vid position 508.
Patogenes
Mutationer i genen för cystisk fibros stör strukturen och funktionen hos ett protein som kallas CFTR (cystisk fibros transmembranregulator). Detta protein fungerar som en kloridkanal som är involverad i vatten-elektrolytutbytet i epitelceller i bronkopulmonala systemet, mag-tarmkanalen, bukspottkörteln, levern och reproduktionssystemet. Som ett resultat av störningar i CFTR-proteinets funktion och struktur ackumuleras kloridjoner Cl⁻ inuti cellen. Detta leder till en förändring av den elektriska potentialen i utsöndringskanalernas lumen, vilket underlättar flödet av stora mängder natriumjoner (Na + ) från kanalens lumen in i cellen och ytterligare förbättrar absorptionen av vatten från det pericellulära utrymmet.
Som ett resultat av dessa förändringar förtjockas utsöndringen av de flesta exokrina körtlar, dess evakuering störs, vilket leder till uttalade sekundära störningar i organ och system, mest uttalade i bronkopulmonala och matsmältningssystemet.
En kronisk inflammatorisk process av varierande intensitet utvecklas i bronkerna, funktionen hos det cilierade epitelet störs kraftigt, sputumet blir mycket visköst, tjockt, mycket svårt att tömma, dess stagnation observeras, bronkiolo- och bronkiektasi bildas, vilka med tiden blir vanligare. Dessa förändringar leder till en ökning av hypoxi och bildandet av kronisk pulmonell hjärtsjukdom.
Patienter med cystisk fibros är extremt predisponerade för utveckling av kronisk inflammation i bronkopulmonala systemet. Detta beror på uttalade störningar i det lokala bronkopulmonala försvarssystemet (minskade nivåer av IgA, interferon, fagocyterande funktion hos alveolära makrofager och leukocyter).
Alveolära makrofager spelar en viktig roll i utvecklingen av kronisk inflammation i bronkopulmonala systemet. De producerar stora mängder IL-8, vilket dramatiskt ökar neutrofilkemotaxi i bronkerna. Neutrofiler ackumuleras i stora mängder i bronkerna och utsöndrar, tillsammans med epitelceller, många proinflammatoriska cytokiner, inklusive IL-1, 8, 6, tumörnekrosfaktor och leukotriener.
En viktig roll i patogenesen av skador på bronkopulmonala systemet spelas också av enzymets höga aktivitet elastas. Man skiljer mellan exogent och endogent elastas. Det första produceras av bakteriefloran (särskilt Pseudomonas aeruginosa), det andra av neutrofila leukocyter. Elastas förstör epitelet och andra strukturella element i bronkerna, vilket bidrar till ytterligare störningar av mukociliär transport och snabb bildning av bronkiektasi.
Neutrofila leukocyter utsöndrar även andra proteolytiska enzymer. Alfa-1-antipyrsin och en sekretorisk hämmare av leukoproteaser motverkar proteolytiska enzymers inverkan och skyddar därför bronkopulmonala systemet från deras skadliga inflytande. Tyvärr undertrycks dock dessa skyddande faktorer hos patienter med cystisk fibros av en betydande mängd neutrofilt proteas.
Alla dessa omständigheter bidrar till att infektionen sprids till bronkopulmonala systemet och att kronisk purulent bronkit utvecklas. Dessutom bör man beakta att det defekta proteinet som kodas av genen för cystisk fibros förändrar bronkialepitelets funktionella tillstånd, vilket gynnar bakteriers vidhäftning till bronkialepitelet, främst Pseudomonas aeruginosa.
Tillsammans med patologin i bronkopulmonala systemet orsakar cystisk fibros också allvarliga skador på bukspottkörteln, magsäcken, tjocktarmen och tunntarmen samt levern.
Symtom cystisk fibros
Cystisk fibros manifesterar sig med olika kliniska symtom. Hos nyfödda kan sjukdomen manifestera sig med mekoniumileus. På grund av brist på eller till och med fullständig frånvaro av trypsin blir mekonium mycket tätt, visköst och ackumuleras i ileocekalregionen. Vidare utvecklas tarmobstruktion, vilket manifesteras av intensiv kräkning med gallblandning, utspänd buk, bristande mekoniumutsöndring, utveckling av peritonitsymptom och snabb ökning av kliniska manifestationer av allvarligt berusningssyndrom. Barnet kan dö under de första dagarna i livet om akut kirurgiskt ingrepp inte utförs.
I mindre allvarliga fall är ett karakteristiskt tecken på cystisk fibros riklig, frekvent avföring, lös, med stor mängd fett och en mycket obehaglig lukt. Hos 1/3 av patienterna observeras ändtarmprolaps.
Därefter fortsätter patienterna att uppleva tarmdysfunktion, malabsorptionssyndrom, allvarliga fysiska utvecklingsstörningar och svår hypovitaminos.
Under det första eller andra levnadsåret uppstår symtom på skador på bronkopulmonala systemet (en mild form av sjukdomen), vilket manifesteras av en hosta som kan vara extremt uttalad och likna hosta med kikhosta. Hostan åtföljs av cyanos, andnöd och separation av tjockt slem, initialt slemhinnigt och sedan varigt. Gradvis bildas en klinisk bild av kronisk obstruktiv bronkit och bronkiektasi, lungemfysem och andningssvikt. Barn är extremt mottagliga för akuta respiratoriska virus- och bakterieinfektioner, vilket bidrar till exacerbationer och progression av bronkopulmonell patologi. Utveckling av infektionsberoende bronkialastma är möjlig.
Hos skolbarn kan cystisk fibros manifestera sig som "tarmkolik". Patienter klagar över svåra paroxysmala buksmärtor, uppblåsthet och upprepade kräkningar. Vid palpering av buken upptäcks täta formationer, belägna i tjocktarmen - avföring blandad med tjockt, tätt slem. Barn är mycket benägna att utveckla hypokloremisk alkalos på grund av överdriven utsöndring av salt med svett i varmt väder, medan "saltfrost" uppträder på barnets hud.
Bronkopulmonala sjukdomar hos vuxna
Skador på bronkopulmonala systemet hos patienter med cystisk fibros (lungformen av sjukdomen) kännetecknas av utveckling av kronisk purulent obstruktiv bronkit, bronkiektasi, kronisk lunginflammation, lungemfysem, andningssvikt och pulmonell hjärtsjukdom. Vissa patienter utvecklar pneumothorax och andra komplikationer av cystisk fibros: atelektas, lungabscesser, hemoptys, lungblödning och infektionsberoende bronkialastma.
Patienter klagar över en smärtsam paroxysmal hosta med mycket viskös, svårseparerad mukopurulent sputum, ibland med en blandning av blod. Dessutom är andnöd extremt karakteristisk, först vid fysisk ansträngning och sedan i vila. Andnöd orsakas av bronkial obstruktion. Många patienter klagar över kronisk rinit orsakad av polypos och bihåleinflammation. Betydande svaghet, progressiv minskning av prestationsförmågan och frekventa akuta luftvägsvirussjukdomar är också karakteristiska. Vid undersökning uppmärksammas blek hud, svullnader i ansiktet, cyanos i synliga slemhinnor och svår andnöd. Med utvecklingen av dekompenserad pulmonell hjärtsjukdom uppträder ödem i benen. Förtjockning av fingrarnas terminala falanger i form av trummor och naglar i form av urglas kan observeras. Bröstkorgen antar en tunnformad form (på grund av utveckling av lungemfysem).
Perkussion av lungorna avslöjar tecken på emfysem - ett boxljud, en skarp begränsning av rörligheten i lungkanten och en sänkning av lungans nedre kant. Auskultation av lungorna avslöjar hård andning med förlängd utandning, spridd torr väsande andning och fuktig medel och fin bubblande väsande andning. Vid svårt lungemfysem försvagas andningen kraftigt.
Extrapulmonära manifestationer av cystisk fibros
Extrapulmonära manifestationer av cystisk fibros kan vara ganska uttalade och förekomma ofta.
Skada på bukspottkörteln
Brist på bukspottkörtelns exokrina funktion av varierande svårighetsgrad observeras hos 85 % av patienter med cystisk fibros. Vid mindre skador på bukspottkörteln saknas dålig matsmältning och malabsorptionssyndrom, det finns endast laboratoriemanifestationer av exokrin insufficiens (låga nivåer av trypsin och lipas i blodet och duodenalinnehållet; ofta svår steatorré). Det är känt att för att förebygga dålig matsmältningssyndrom räcker det med en utsöndring av endast 1 till 2 % av det totala lipaset. Endast signifikanta störningar i den exokrina funktionen manifesteras kliniskt.
Under normala förhållanden producerar bukspottkörtelns acini ett flytande sekret rikt på enzymer. När sekretet rör sig längs utsöndringsgången berikas det med vatten och anjoner, och det blir ännu mer flytande. Vid cystisk fibros, på grund av en störning i transmembranregulatorns (kloridkanalens) struktur och funktion, får inte bukspottkörtelns sekret en tillräcklig mängd vätska, det blir visköst och hastigheten på dess rörelse längs utsöndringsgången minskar kraftigt. Sekretproteinerna avsätts på väggarna i små utsöndringsgångar, vilket resulterar i deras blockering. Allt eftersom sjukdomen fortskrider utvecklas slutligen förstörelse och atrofi av acini - kronisk pankreatit med exokrin pankreasinsufficiens bildas. Detta återspeglas kliniskt i utvecklingen av matsmältningsproblem och malabsorptionssyndrom. Bukspottkörtelinsufficiens är den främsta orsaken till fettmalabsorption vid cystisk fibros, men detta observeras vanligtvis vid en signifikant lipasbrist. Forsher och Durie (1991) indikerar att i fullständig avsaknad av pankreaslipas bryts fett ner och absorberas med 50-60%, vilket beror på närvaron av mag- och salivlipaser (sublinguala), vars aktivitet ligger nära den nedre gränsen för normen. Tillsammans med störningen av fettnedbrytning och absorption sker en störning av proteinnedbrytning och reabsorption. Cirka 50% av proteinet som intas med mat förloras med avföringen. Kolhydratabsorptionen påverkas i mindre utsträckning trots brist på α-amylas, men kolhydratmetabolismen kan störas avsevärt.
Skador på bukspottkörteln uttrycks i utvecklingen av matsmältningsproblem och malabsorptionssyndrom med betydande viktminskning och riklig fet avföring.
Utvecklingen av matsmältningsproblem och malabsorptionssyndrom underlättas också av allvarlig dysfunktion i tarmkörtlarna, nedsatt utsöndring av tarmsaft och en minskning av innehållet av tarmenzymer i den.
Maldigestion- och malabsorptionssyndrom kallas också den intestinala formen av cystisk fibros.
Nedsatt endokrin funktion i bukspottkörteln (diabetes mellitus) observeras hos patienter med cystisk fibros i sjukdomens sena skeden (hos 2 % av barnen och 15 % av vuxna)
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Lever- och gallvägsskador
Hos 13 % av patienter med blandade och intestinala former av cystisk fibros utvecklas levercirros. Det är mest typiskt för mutationerna W128X, delta-P508 och X1303K. Gallvägscirros med portalhypertension detekteras hos 5–10 % av patienterna. Enligt Welch, Smith (1995) detekteras kliniska, morfologiska, laboratoriemässiga och instrumentella tecken på leverskada hos 86 % av patienter med cystisk fibros.
Många patienter med cystisk fibros utvecklar också kronisk kolecystit, ofta kalkfri.
Dysfunktion i könskörtlarna
Patienter med cystisk fibros kan uppleva azoospermi, vilket är orsaken till infertilitet. Minskad fertilitet är också typiskt för kvinnor.
Stages
Det finns tre svårighetsgrader av pulmonell cystisk fibros.
Den milda formen av cystisk fibros kännetecknas av sällsynta exacerbationer (högst en gång per år); under perioder av remission är kliniska manifestationer praktiskt taget frånvarande och patienterna kan arbeta.
Måttlig svårighetsgrad - exacerbationer observeras 2-3 gånger per år och varar cirka 2 månader eller längre. I exacerbationsfasen förekommer intensiv hosta med svår att separera slem, andnöd även vid mindre fysisk ansträngning, subfebril kroppstemperatur, allmän svaghet, svettningar. Samtidigt finns det en kränkning av bukspottkörtelns exokrina funktion. I remissionsfasen återställs inte arbetsförmågan helt, andnöden vid fysisk ansträngning kvarstår.
Svårt förlopp kännetecknas av mycket frekventa exacerbationer av sjukdomen. Remissioner är praktiskt taget frånvarande. I den kliniska bilden framträder allvarlig andningssvikt, symtom på kronisk pulmonell hjärtsjukdom, ofta dekompenserad, hemoptys är typisk, i förgrunden. Signifikant viktminskning observeras, patienterna är helt funktionsnedsatta. Som regel åtföljs allvarlig bronkopulmonell patologi av en kraftigt uttalad dysfunktion i bukspottkörteln.
Formulär
- Bronkopulmonala lesioner
- Upprepad och återkommande lunginflammation med utdraget förlopp.
- Abscesserande lunginflammation, särskilt hos spädbarn.
- Kronisk lunginflammation, särskilt bilateral.
- Bronkial astma som är refraktär mot traditionell behandling.
- Återkommande bronkit, bronkiolit, särskilt vid Pseudomonas aeruginosa-odling.
- Förändringar i mag-tarmkanalen
- Meconium ileus och dess motsvarigheter.
- Syndrom med nedsatt intestinal absorption av okänd genes.
- Obstruktiv gulsot hos nyfödda med ett utdraget förlopp.
- Levercirros.
- Diabetes mellitus.
- Gastroesofageal reflux.
- Kolelitiasis.
- Rektal prolaps.
- Förändringar i andra organ och system
- Tillväxt- och utvecklingsstörningar.
- Försenad sexuell utveckling.
- Manlig infertilitet.
- Näspolyper.
- Syskon från familjer med cystisk fibros.
[ 24 ]
Komplikationer och konsekvenser
Komplikationer från mag-tarmkanalen inkluderar:
- Diabetes mellitus utvecklas hos 8–12 % av patienter över 25 år.
- Fibroserande kolonopati.
- Mekoniumileus under nyföddhetsperioden (hos 12 % av nyfödda med cystisk fibros), distalt tarmobstruktionssyndrom, rektal prolaps, magsår och gastroesofageal refluxsjukdom.
Leverkomplikationer:
- Fettleversjukdom (hos 30–60 % av patienterna),
- Fokal biliär cirros, multinodulär biliär cirros och associerad portalhypertension.
Portalhypertension leder ibland till döden på grund av esofagusvaricer.
Förekomsten av kolecystit och gallsten är högre hos patienter med cystisk fibros än hos andra individer.
Försenad pubertet och minskad fertilitet och andra komplikationer. De flesta män har azoospermi och underutveckling av sädesledaren.
Diagnostik cystisk fibros
Allmän blodanalys - anemi av varierande svårighetsgrad är typisk, vanligtvis normo- eller hypokrom. Anemi har en polyfaktoriell genes (minskat absorption av järn och vitamin B12 i tarmen på grund av utveckling av malabsorptionssyndrom). Leukopeni är möjlig, med utveckling av purulent bronkit och lunginflammation - leukocytos, ökad ESR.
Allmän urinanalys - inga signifikanta förändringar, i sällsynta fall observeras lätt proteinuri.
Koprologisk undersökning - steatorré och kreatorré observeras. Becker (1987) rekommenderar mätning av kymotrypsin och fettsyror i avföringen. Innan kymotrypsin i avföringen bestäms är det nödvändigt att sluta ta matsmältningsenzymer minst 3 dagar före undersökningen. Vid cystisk fibros minskar mängden kymotrypsin i avföringen och mängden fettsyror ökar (normal utsöndring av fettsyror är mindre än 20 mmol/dag). Det är nödvändigt att ta hänsyn till att ökad utsöndring av fettsyror med avföringen också observeras vid:
- brist på konjugerade fettsyror i tunntarmen på grund av leversvikt, obstruktion av gallgångarna, betydande bakteriell kolonisering av tunntarmen (i detta fall sker intensiv hydrolys av gallsyror);
- ileit;
- celiaki (med utveckling av malabsorptionssyndrom);
- enterit;
- intestinala lymfom;
- Whipples sjukdom;
- matallergier;
- accelererad transit av matmassor vid diarré av olika ursprung, karcinoidsyndrom, tyreotoxikos.
Biokemiskt blodprov - minskade nivåer av totalt protein och albumin, förhöjda nivåer av alfa2- och gammaglobuliner, bilirubin och aminotransferaser (vid leverskada), minskad aktivitet av amylas, lipas, trypsin samt järn- och kalciumnivåer (vid utveckling av matsmältningsproblem, malabsorption).
Sputumanalys - närvaro av ett stort antal neutrofila leukocyter och mikroorganismer (under sputumbakterioskopi).
En studie av tunntarmens absorptionsfunktion och bukspottkörtelns exokrina funktion avslöjar betydande störningar.
Röntgenundersökning av lungorna - avslöjar förändringar, vars svårighetsgrad beror på sjukdomens svårighetsgrad och fas. De mest karakteristiska förändringarna är:
- ökad lungmönstring på grund av peribronkiala interstitiella förändringar;
- expansion av lungrötterna;
- bild av lobulär, subsegmental eller till och med segmental atelektas i lungorna;
- ökad transparens i lungfälten, främst i de övre delarna, låg position och otillräcklig rörlighet hos diafragman, expansion av det retrosternala utrymmet (manifestation av lungemfysem);
- segmental eller polysegmental infiltration av lungvävnad (vid utveckling av lunginflammation).
Bronkografi avslöjar förändringar orsakade av obstruktion av bronkierna av visköst sputum (fragmentering av bronkiernas fyllning med kontrastmedel, ojämna konturer, fenomenet bronkial bristning, en signifikant minskning av antalet laterala grenar), samt bronkoekgaser (cylindriska, blandade), lokaliserade huvudsakligen i lungornas nedre delar.
Bronkoskopi avslöjar diffus varig bronkit med riklig mängd tjock, viskös sputum och fibrinösa filmer.
Spirometri - redan i sjukdomens tidiga stadier avslöjar andningssvikt av obstruktiv typ (minskat FVC, FEV1, Tiffno-index), restriktiv (minskat FVC) eller, oftast, obstruktiv-restriktiv (minskat FVC, FVC, FEV1, Tiffno-index).
Gibson och Cooks svetttest (svettelektrolyttest) innebär att stimulera svettning med hjälp av pilokarpinelektrofores med efterföljande bestämning av klorider i svetten. Doerehuk (1987) beskriver testet enligt följande. Pilokarpinelektrofores utförs på underarmen, den elektriska strömmen är 3 mA. Efter rengöring av huden med destillerat vatten samlas svetten upp med hjälp av filterpapper som placeras på det stimulerade området, täckt med gasbinda för att förhindra avdunstning från det. Efter 30-60 minuter avlägsnas filterpappret och elueras i destillerat vatten. Mängden uppsamlad svett mäts. För att få tillförlitliga resultat är det nödvändigt att samla upp minst 50 mg (helst 100 mg) svett.
Om kloridkoncentrationen är mer än 60 mmol/l anses diagnosen cystisk fibros vara sannolik; om kloridkoncentrationen är mer än 100 mmol/l är den tillförlitlig; i detta fall bör skillnaden i koncentrationen av klor och natrium inte överstiga 8-10 mmol/l. Hadson (1983) rekommenderar att om natrium- och kloridhalten i svett är gränsfallande bör ett prednisolontest utföras (5 mg oralt i 2 dagar, följt av bestämning av elektrolyter i svett). Hos individer som inte lider av cystisk fibros minskar natriumnivån i svett till den nedre normalgränsen; vid cystisk fibros förändras den inte. Ett svetttest rekommenderas för alla barn med kronisk hosta.
Analys av blodfläckar eller DNA-prover för större mutationer i genen för cystisk fibros är det känsligaste och mest specifika diagnostiska testet. Denna metod är dock endast lämplig för länder där mutationsfrekvensen för delta-P508 är högre än 80 %. Dessutom är tekniken mycket dyr och tekniskt komplex.
Prenatal diagnos av cystisk fibros utförs genom att bestämma isoenzymerna av alkaliskt fosfatas i fostervätska. Denna metod är möjlig från 18-20 veckors graviditet.
De viktigaste kriterierna för att diagnostisera cystisk fibros är följande:
- indikationer i anamnesen på försenad fysisk utveckling i barndomen, återkommande kroniska luftvägssjukdomar, dyspeptiska störningar och diarré, förekomst av cystisk fibros hos nära släktingar;
- kronisk obstruktiv bronkit, ofta återkommande, med utveckling av bronkiektasi och lungemfysem, ofta återkommande lunginflammation;
- kronisk återkommande pankreatit med en markant minskning av exokrin funktion, malabsorptionssyndrom;
- ökat klorinnehåll i patientens svett;
- infertilitet med bibehållen sexuell funktion.
Framgångsrik diagnos och differentialdiagnos av cystisk fibros underlättas genom att riskgrupper identifieras.
Screeningprogram för cystisk fibros
- Allmän analys av blod, urin, sputum.
- Bakteriologisk analys av sputum.
- Koprologisk analys.
- Biokemiskt blodprov: bestämning av totalt protein och proteinfraktioner, glukos, bilirubin, aminotransferaser, alkaliskt fosfatas, gamma-glutamyltranspeptidaser, kalium, kalcium, järn, lipas, amylas, trypsin.
- Studie av bukspottkörtelns exokrina funktion och tarmens absorptionsfunktion.
- Fluoroskopi och röntgen av lungorna, datortomografi av lungorna.
- EKG.
- Ekokardiografi.
- Bronkoskopi och bronkografi.
- Spirometri.
- Svetttest.
- Konsultation med en genetiker.
- Analys av blodfläckar eller DNA-prover för större mutationer i genen för cystisk fibros.
Vad behöver man undersöka?
Hur man undersöker?
Vilka tester behövs?
Vem ska du kontakta?
Behandling cystisk fibros
Typen och svårighetsgraden av symtom på cystisk fibros kan variera kraftigt, så det finns ingen typisk behandlingsplan; den är individualiserad för varje individ.
Terapin består av följande terapeutiska åtgärder:
- Andningsövningar och postural dränering hjälper till att bli av med tjockt slem som samlas i lungorna. Vissa tekniker för att rensa luftvägarna kräver hjälp från familjemedlemmar, vänner eller en lungspecialist. Många använder en uppblåsbar bröstväst som vibrerar med hög frekvens.

- Inhalationsläkemedel som har bronkvidgande, dränerande (mukolytiska) och antibakteriella effekter (till exempel fluorokinoloner).
- Preparat som innehåller pankreatiska enzymer för att förbättra matsmältningen. Dessa preparat tas i samband med måltider.
- Multivitaminer (inklusive fettlösliga vitaminer).
År 2015 godkände FDA ett andra läkemedel för behandling av cystisk fibros som riktar sig mot ett defekt protein som kallas CFTR. Det första läkemedlet, en så kallad CFTR-modulator, godkändes 2012. CFTR-modulatorer förväntas förlänga livet för vissa personer med cystisk fibros med årtionden.
Kirurgi kan krävas för att behandla följande andningskomplikationer:
- Pneumothorax, massiv återkommande eller ihållande hemoptys, näspolyper, ihållande och kronisk bihåleinflammation.
- Mekoniumileus, invagination, rektal prolaps.
Lungtransplantation utförs i sjukdomens terminala skede.
Prognos
Den genomsnittliga överlevnadsåldern för patienter med cystisk fibros varierar från 35 till 40 år. Den genomsnittliga överlevnadsåldern är högre för män än för kvinnor.
Med moderna behandlingsstrategier når 80 % av patienterna vuxen ålder. Cystisk fibros begränsar dock patientens funktionella förmåga avsevärt. Det finns fortfarande inget botemedel mot denna sjukdom.