Medicinsk expert av artikeln

Nya publikationer
Ärftlig nefrit (Alports syndrom) hos barn
Senast recenserade: 05.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
Ärftlig nefrit (Alports syndrom) är en genetiskt betingad ärftlig icke-immun glomerulopati, som manifesteras av hematuri (ibland med proteinuri), progressiv nedsättning av njurfunktionen med utveckling av kronisk njursvikt, ofta i kombination med sensorineural dövhet och synnedsättning.
Sjukdomen beskrevs först 1902 av LG Guthrie, som observerade en familj där hematuri observerades i flera generationer. År 1915 beskrev A.F. Hurst utvecklingen av uremi hos medlemmar i samma familj. År 1927 identifierade A. Alport först hörselnedsättning hos flera släktingar med hematuri. På 1950-talet beskrevs ögonskador vid en liknande sjukdom. År 1972, hos patienter med ärftlig hematuri, under en morfologisk studie av njurvävnad, avslöjade Hinglais et al. ojämn expansion och stratifiering av glomerulära basalmembran. År 1985 identifierades den genetiska grunden för ärftlig nefrit - en mutation i typ IV-kollagengenen (Fiengold et al., 1985).
Studien av sjukdomens genetiska natur gjorde det möjligt att dra slutsatsen att skillnaderna i de fenotypiska manifestationerna av ärftlig nefrit (med eller utan hörselnedsättning) beror på graden av uttryck av den muterade genen. Således betraktas för närvarande alla kliniska varianter som manifestationer av en sjukdom och termen "ärftlig nefrit" är synonym med termen "Alports syndrom".
Enligt epidemiologiska studier förekommer ärftlig nefrit med en frekvens av 17 per 100 000 barn.
 [
[ Orsaker till Alports syndrom
Den genetiska grunden för sjukdomen är en mutation i genen för a-5-kedjan av typ IV-kollagen. Denna typ är universell för basalmembranen i njuren, cochleaapparaten, linskapseln, näthinnan och hornhinnan i ögat, vilket har bevisats i studier med monoklonala antikroppar mot denna kollagenfraktion. Nyligen har möjligheten att använda DNA-prober för prenatal diagnostik av ärftlig nefrit indikerats.
Vikten av att testa alla familjemedlemmar med DNA-sonder för att identifiera bärare av den muterade genen betonas, vilket är av stor betydelse för att genomföra medicinsk och genetisk rådgivning av familjer med denna sjukdom. Emellertid har upp till 20 % av familjerna inga släktingar som lider av njursjukdom, vilket tyder på en hög frekvens av spontana mutationer av den onormala genen. De flesta patienter med ärftlig nefrit har individer med njursjukdom, hörselnedsättning och synsjukdom i sina familjer; blodsäktenskap mellan personer med en eller flera förfäder är viktiga, eftersom sannolikheten att få samma gener från båda föräldrarna ökar i äktenskap mellan besläktade individer. Autosomalt dominanta, autosomalt recessiva och dominanta, X-länkade överföringsvägar har etablerats.
Hos barn skiljer man oftast åt i tre typer av ärftlig nefrit: Alports syndrom, ärftlig nefrit utan hörselnedsättning och familjär benign hematuri.
Alports syndrom är en ärftlig nefrit med hörselnedsättning. Den är baserad på en kombinerad defekt i kollagenstrukturen i det glomerulära basalmembranet i njurarna, örat och ögonstrukturerna. Genen för klassiskt Alports syndrom är belägen i locus 21-22q på X-kromosomens långa arm. I de flesta fall ärvs det på ett dominant sätt, kopplat till X-kromosomen. I detta avseende är Alports syndrom allvarligare hos män, eftersom hos kvinnor kompenseras den muterade genens funktion av en frisk allel av den andra, oskadade kromosomen.
Den genetiska grunden för utvecklingen av ärftlig nefrit är mutationer i generna för alfa-kedjorna i typ IV-kollagen. Sex alfa-kedjor av typ IV-kollagen G är kända: generna för a5- och a6-kedjorna (Col4A5 och Col4A5) är belägna på X-kromosomens långa arm i 21-22q-zonen; generna för a3- och a4-kedjorna (Col4A3 och Col4A4) är belägna på den andra kromosomen; generna för a1- och a2-kedjorna (Col4A1 och Col4A2) är belägna på den 13:e kromosomen.
I de flesta fall (80–85 %) upptäcks ett X-länkat nedärvningsmönster av sjukdomen, associerat med skador på Col4A5-genen till följd av deletion, punktmutationer eller splitsningsrubbningar. För närvarande har mer än 200 mutationer av Col4A5-genen hittats, vilka är ansvariga för störningen av syntesen av a5-kedjorna av typ IV-kollagen. Vid denna typ av nedärvning manifesterar sig sjukdomen hos barn av båda könen, men hos pojkar är den allvarligare.
Mutationer i loci för Col4A3- och Col4A4-generna som ansvarar för syntesen av a3- och a4-kedjorna av typ IV-kollagen ärvs autosomalt. Enligt forskning observeras den autosomalt dominanta typen av nedärvning i 16 % av fallen av ärftlig nefrit, och den autosomalt recessiva typen observeras hos 6 % av patienterna. Cirka 10 varianter av mutationer av Col4A3- och Col4A4-generna är kända.
Resultatet av mutationer är en kränkning av sammansättningsprocesserna för typ IV-kollagen, vilket leder till en kränkning av dess struktur. Typ IV-kollagen är en av huvudkomponenterna i det glomerulära basalmembranet, cochleaapparaten och ögats lins, vars patologi kommer att detekteras i kliniken för ärftlig nefrit.
Kollagen typ IV, som är en del av det glomerulära basalmembranet, består huvudsakligen av två a1-kedjor (IV) och en a2-kedja (IV), och innehåller även a3-, a4-, a5-kedjor. Oftast, vid X-länkad nedärvning, åtföljs mutationen av Col4A5-genen av frånvaron av a3-, a4-, a5- och a6-kedjor i strukturen av kollagen typ IV, och antalet o1- och a2-kedjor i det glomerulära basalmembranet ökar. Mekanismen för detta fenomen är oklar, det antas att orsaken är posttranskriptionella förändringar i mRNA.
Avsaknaden av a3-, a4- och a5-kedjor i strukturen av typ IV-kollagen i glomerulära basalmembran leder till deras förtunning och skörhet i de tidiga stadierna av Alports syndrom, vilket kliniskt manifesteras oftare av hematuri (mindre ofta av hematuri med proteinuri eller endast proteinuri), hörselnedsättning och lenticonus. Ytterligare progression av sjukdomen leder till förtjockning och nedsatt permeabilitet hos basalmembranen i de sena stadierna av sjukdomen, med proliferation av kollagen av typ V och VI i dem, vilket manifesteras i en ökning av proteinuri och en minskning av njurfunktionen.
Mutationens natur som ligger bakom ärftlig nefrit avgör i hög grad dess fenotypiska manifestation. Vid deletion av X-kromosomen med samtidig mutation av Col4A5- och Col4A6-generna som ansvarar för syntesen av a5- och a6-kedjorna av typ IV-kollagen kombineras Alports syndrom med leiomyomatos i matstrupen och könsorganen. Enligt forskningsdata noteras vid en mutation av Col4A5-genen i samband med en deletion en större svårighetsgrad av den patologiska processen, en kombination av njurskada med extrarenala manifestationer och tidig utveckling av kronisk njursvikt, jämfört med en punktmutation av denna gen.
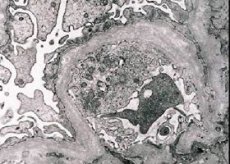
Morfologiskt sett avslöjar elektronmikroskopi förtunning och stratifiering av glomerulära basalmembran (särskilt lamina densa) och förekomsten av elektrontäta granuler. Glomerulära lesioner kan vara heterogena hos samma patient, från minimala fokala mesangiala lesioner till glomeruloskleros. Glomerulit vid Alports syndrom är alltid immunonegativ, vilket skiljer den från glomerulonefrit. Karakteristiska drag inkluderar utveckling av tubulär atrofi, lymfohistiocytisk infiltration och förekomsten av "skumceller" med lipidinklusioner - lipofager. Allt eftersom sjukdomen fortskrider avslöjas förtjockning och uttalad destruktion av glomerulära basalmembran.
Vissa förändringar i immunsystemet avslöjas. Patienter med ärftlig nefrit har en minskad IgA-nivå och en tendens att öka IgM-koncentrationen i blodet. IgG-nivån kan vara förhöjd i sjukdomens tidiga stadier och minska i senare stadier. Kanske är ökningen av IgM- och G-koncentrationen en slags kompensationsreaktion som svar på IgA-brist.
T-lymfocytsystemets funktionella aktivitet minskar; en selektiv minskning av B-lymfocyter som är ansvariga för syntesen av Ig A noteras, den fagocytiska länken till immunitet störs, främst på grund av störningar i kemotaxi och intracellulära matsmältningsprocesser i neutrofiler.
Vid undersökning av njurbiopsi hos patienter med Alports syndrom avslöjar elektronmikroskopidata ultrastrukturella förändringar i glomerulärt basalmembran: förtunning, strukturstörningar och delning av glomerulära basalmembran med en förändring i dess tjocklek och ojämna konturer. I de tidiga stadierna av ärftlig nefrit bestämmer defekten förtunningen och skörheten hos glomerulära basalmembran.
Förtunning av glomerulära membran är ett mer gynnsamt tecken och är vanligare hos flickor. Ett mer konstant elektronmikroskopiskt tecken vid ärftlig nefrit är delning av basalmembranet, och svårighetsgraden av dess förstörelse korrelerar med processens svårighetsgrad.
Symtom på Alports syndrom hos barn
De första symptomen på Alports syndrom i form av isolerat urinsyndrom upptäcks oftast hos barn under de första tre åren av livet. I de flesta fall upptäcks sjukdomen av en slump. Urinsyndrom upptäcks under en förebyggande undersökning av barnet, före inläggning på en förskola eller under ARVI. Vid patologi i urinen under ARVI. Vid ärftlig nefrit, till skillnad från förvärvad glomerulonefrit, finns ingen latent period.
I sjukdomens inledande skede lider barnets hälsa lite, ett karakteristiskt drag är urinsyndromets ihållande och motståndskraft. Ett av huvudtecknen är hematuri av varierande svårighetsgrad, observerat i 100% av fallen. En ökning av graden av hematuri noteras under eller efter luftvägsinfektioner, fysisk aktivitet eller efter förebyggande vaccinationer. Proteinuri överstiger i de flesta fall inte 1 g/dag, i början av sjukdomen kan vara oregelbunden, allt eftersom processen fortskrider ökar proteinuri. Periodiskt kan leukocyturi med en övervikt av lymfocyter förekomma i urinsedimentet, vilket är förknippat med utvecklingen av interstitiella förändringar.
Därefter försämras partiell njurfunktion, patientens allmäntillstånd försämras: berusning, muskelsvaghet, arteriell hypotoni, ofta hörselnedsättning (särskilt hos pojkar) och ibland synnedsättning uppstår. Berusning manifesteras av blekhet, trötthet och huvudvärk. I sjukdomens inledande skede upptäcks hörselnedsättning i de flesta fall endast med audiografi. Hörselnedsättning vid Alports syndrom kan uppstå under olika perioder i barndomen, men oftast diagnostiseras hörselnedsättning vid 6-10 års ålder. Hörselnedsättning hos barn börjar med höga frekvenser, når en betydande grad i luft- och benledning, och övergår från ljudledande till ljuduppfattande hörselnedsättning. Hörselnedsättning kan vara ett av de första symtomen på sjukdomen och kan föregå urinvägssyndrom.
I 20 % av fallen har patienter med Alports syndrom förändringar i synorganen. De vanligaste avvikelserna är linsens: sfärofoki, främre, bakre eller blandad lenticonus, och olika grå starr. I familjer med Alports syndrom finns en betydande frekvens av myopi. Ett antal forskare noterar ständigt bilaterala perimakulära förändringar i dessa familjer i form av ljusa vitaktiga eller gulaktiga granuleringar i corpus luteum. De anser att detta tecken är ett konstant symptom som har högt diagnostiskt värde vid Alports syndrom. KS Chugh et al. (1993) fann i en oftalmologisk studie hos patienter med Alports syndrom en minskning av synskärpan i 66,7 % av fallen, främre lenticonus i 37,8 %, retinala fläckar i 22,2 %, grå starr i 20 % och keratokonus i 6,7 %.
Hos vissa barn med ärftlig nefrit, särskilt vid njursvikt, noteras en betydande eftersläpning i den fysiska utvecklingen. Allt eftersom njursvikten fortskrider utvecklas arteriell hypertoni. Hos barn upptäcks det oftare i tonåren och i äldre åldersgrupper.
Patienter med ärftlig nefrit kännetecknas av förekomsten av olika (mer än 5-7) stigmat som tyder på bindvävsdysmorfogenes. Bland bindvävsstigmat hos patienter är de vanligaste hypertelorism i ögonen, hög gom, bettanomalier, onormal form på öronen, krökning av lillfingret på händerna och "sandalgap" på fötterna. Ärftlig nefrit kännetecknas av enhetligheten av dysmorfogenesstigmat inom en familj, samt en hög frekvens av deras spridning bland släktingar till probander längs vars linje sjukdomen överförs.
I sjukdomens tidiga stadier upptäcks en isolerad minskning av partiella njurfunktioner: transport av aminosyror, elektrolyter, koncentrationsfunktion, acidogenes, senare förändringar påverkar funktionstillståndet hos både den proximala och distala delen av nefronet och kännetecknas av kombinerade partiella störningar. En minskning av glomerulär filtration sker senare, oftare i tonåren. När den ärftliga nefriten fortskrider utvecklas anemi.
Således kännetecknas ärftlig nefrit av ett stegvis förlopp av sjukdomen: först ett latent stadium eller dolda kliniska symtom, manifesterade av minimala förändringar i urinsyndromet, sedan sker en gradvis dekompensation av processen med en minskning av njurfunktionen med tydliga kliniska symtom (intoxikation, asteni, utvecklingsförsening, anemi). Kliniska symtom uppträder vanligtvis oavsett skiktningen av den inflammatoriska reaktionen.
Ärftlig nefrit kan manifestera sig vid olika åldersperioder, vilket beror på genens verkan, som är i ett undertryckt tillstånd fram till en viss tidpunkt.
Klassificering
Det finns tre typer av ärftlig nefrit
- Alternativ I - manifesterar sig kliniskt som nefrit med hematuri, hörselnedsättning och ögonskador. Nefritens förlopp är progressivt med utvecklingen av kronisk njursvikt. Arvstypen är dominant, kopplad till X-kromosomen. Morfologiskt avslöjas en kränkning av basalmembranets struktur, dess uttunning och delning.
- Alternativ II - manifesterar sig kliniskt som nefrit med hematuri utan hörselnedsättning. Nefritens förlopp är progressivt med utveckling av kronisk njursvikt. Arvstypen är dominant, kopplad till X-kromosomen. Morfologiskt detekteras en förtunning av det glomerulära kapillärbasalmembranet (särskilt laminadensa).
- Alternativ III - benign familjär hematuri. Förloppet är gynnsamt, kronisk njursvikt utvecklas inte. Arvstypen är autosomalt dominant eller autosomalt recessiv. Vid autosomalt recessiv arvstyp noteras ett svårare sjukdomsförlopp hos kvinnor.
Diagnos av Alports syndrom
Följande kriterier föreslås:
- förekomsten av minst två patienter med nefropati i varje familj;
- hematuri som det ledande symptomet på nefropati hos probanden;
- förekomsten av hörselnedsättning hos minst en familjemedlem;
- utveckling av kronisk njursvikt hos en eller flera släktingar.
Vid diagnostik av olika ärftliga och medfödda sjukdomar ges stor plats åt en omfattande undersökningsmetod och framför allt åt att uppmärksamma de data som erhållits vid sammanställning av barnets stamtavla. Diagnosen Alports syndrom anses giltig i fall där 3 av 4 typiska tecken upptäcks hos patienten: förekomst av hematuri och kronisk njursvikt i familjen, förekomst av neurosensorisk hörselnedsättning, synpatologi hos patienten, upptäckt av tecken på klyvning av glomerulärt basalmembran med en förändring i dess tjocklek och ojämna konturer under elektronmikroskopiska egenskaper hos biopsin.
Patientens undersökning bör omfatta kliniska och genetiska forskningsmetoder; riktad studie av sjukdomshistorien; allmän undersökning av patienten med hänsyn till diagnostiskt signifikanta kriterier. I kompensationsstadiet kan patologi endast upptäckas genom att fokusera på sådana syndrom som förekomsten av en ärftlig belastning, hypotoni, multipla stigman för dysembryogenes, förändringar i urinsyndromet. I dekompensationsstadiet kan extrarenala symtom uppstå, såsom svår berusning, asteni, försenad fysisk utveckling, anemi, som manifesterar och intensifieras med en gradvis minskning av njurfunktionen. Hos de flesta patienter, med en minskning av njurfunktionen observeras följande: minskad acido- och aminogenes; 50% av patienterna noterar en signifikant minskning av njurarnas sekretoriska funktion; begränsat intervall av fluktuationer i urinens optiska densitet; störning av filtreringsrytmen och sedan en minskning av glomerulär filtration. Stadiet av kronisk njursvikt diagnostiseras när patienter har en förhöjd nivå av urea i blodserumet (mer än 0,35 g/l) i 3-6 månader eller mer, och en minskning av glomerulär filtration till 25% av normen.
Differentialdiagnostik av ärftlig nefrit bör i första hand utföras vid den hematuriska formen av förvärvad glomerulonefrit. Förvärvad glomerulonefrit har oftast en akut debut, en period av 2-3 veckor efter en infektion, extrarenala tecken, inklusive hypertoni från de första dagarna (vid ärftlig nefrit tvärtom hypotoni), minskad glomerulär filtration vid sjukdomsdebut, ingen försämring av partiella tubulära funktioner, medan de vid ärftlig förekommer. Förvärvad glomerulonefrit uppträder med mer uttalad hematuri och proteinuri, med ökad ESR. Typiska förändringar i glomerulärt basalmembran, karakteristiska för ärftlig nefrit, är av diagnostiskt värde.
Differentialdiagnostik vid dysmetabolisk nefropati utförs vid kronisk njursvikt, i familjen kliniskt upptäckta heterogena njursjukdomar, och det kan finnas ett spektrum av nefropati från pyelonefrit till urolithiasis. Barn har ofta klagomål på smärtor i buken och periodvis vid urinering, i urinsedimentet - oxalater.
Vid misstanke om ärftlig nefrit bör patienten remitteras till en specialiserad nefrologisk avdelning för att klargöra diagnosen.
Vad behöver man undersöka?
Hur man undersöker?
Vilka tester behövs?
Vem ska du kontakta?
Behandling av Alports syndrom
Kuren inkluderar begränsningar av hård fysisk ansträngning och exponering för frisk luft. Kosten är komplett, med tillräckliga nivåer av kompletta proteiner, fetter och kolhydrater, med hänsyn till njurfunktionen. Av stor vikt är upptäckt och behandling av kroniska infektionsfokus. Följande läkemedel används: ATP, kokarboxylas, pyridoxin (upp till 50 mg/dag), karnitinklorid. Kurer ges 2-3 gånger per år. Vid hematuri förskrivs örtmedicin - brännässla, aroniajuice, rölleka.
Det finns rapporter i utländsk och inhemsk litteratur om behandling med prednisolon och användning av cytostatika. Det är dock svårt att bedöma effekten.
Vid kronisk njursvikt används hemodialys och njurtransplantation.
Det finns inga metoder för specifik (effektiv patogenetisk) behandling för ärftlig nefrit. Alla behandlingsåtgärder syftar till att förebygga och bromsa nedgången i njurfunktionen.
Kosten bör vara balanserad och kaloririk, med hänsyn till njurarnas funktionella tillstånd. I avsaknad av funktionella störningar bör barnets kost innehålla tillräckligt med proteiner, fetter och kolhydrater. Vid tecken på njurfunktionsnedsättning bör mängden protein, kolhydrater, kalcium och fosfor begränsas, vilket fördröjer utvecklingen av kronisk njursvikt.
Fysisk aktivitet bör begränsas; barn rekommenderas att undvika sport.
Kontakt med smittsamma patienter bör undvikas, risken för att utveckla akuta luftvägssjukdomar bör minskas. Sanering av områden med kronisk infektion är nödvändig. Förebyggande vaccinationer utförs inte för barn med ärftlig nefrit, vaccination är endast möjlig vid epidemiologiska indikationer.
Hormonell och immunsuppressiv behandling vid ärftlig nefrit är ineffektiv. Det finns indikationer på viss positiv effekt (minskning av proteinuri och bromsning av sjukdomsprogression) vid långvarig flerårig användning av ciklosporin A och ACE-hämmare.
Vid behandling av patienter används läkemedel som förbättrar ämnesomsättningen:
- pyridoxin - 2–3 mg/kg/dag i 3 doser i 4 veckor;
- kokarboxylas - 50 mg intramuskulärt varannan dag, totalt 10-15 injektioner;
- ATP - 1 ml intramuskulärt varannan dag, 10-15 injektioner;
- vitamin A - 1000 IE/år/dag i 1 dos i 2 veckor;
- E-vitamin - 1 mg/kg/dag i 1 dos i 2 veckor.
Denna typ av terapi hjälper till att förbättra patienternas allmänna tillstånd, minska tubulära dysfunktioner och utförs i kurer 3 gånger om året.
Levamisol kan användas som immunmodulator - 2 mg/kg/dag 2–3 gånger i veckan med 3–4 dagars uppehåll mellan doserna.
Enligt forskningsdata har hyperbarisk syresättning en positiv effekt på svårighetsgraden av hematuri och njurfunktionsnedsättning.
Den mest effektiva metoden för behandling av ärftlig nefrit är snabb njurtransplantation. I detta fall sker inget återfall av sjukdomen vid transplantationen; i en liten andel av fallen (cirka 5 %) kan nefrit utvecklas i den transplanterade njuren i samband med antigener mot glomerulärt basalmembran.
En lovande inriktning är prenatal diagnostik och genteknik. Djurförsök visar hög effektivitet i att överföra normala gener som ansvarar för syntesen av typ IV-kollagen-alfa-kedjor till njurvävnad, varefter syntesen av normala kollagenstrukturer observeras.
Prognos
Prognosen för ärftlig nefrit är alltid allvarlig.
Prognostiskt ogynnsamma kriterier för förloppet av ärftlig nefrit är:
- manligt kön;
- tidig utveckling av kronisk njursvikt hos familjemedlemmar;
- proteinuri (mer än 1 g/dag);
- förtjockning av glomerulära basalmembran enligt mikroskopi;
- akustisk neurit;
- deletion i Col4A5-genen.
Prognosen för benign familjär hematuri är mer gynnsam.
Использованная литература