Medicinsk expert av artikeln

Nya publikationer
Subakut nekrotiserande Leahs encefalomyopati
Senast recenserade: 04.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
 [
[ Orsaker av Leahs syndrom
Sjukdomen beror på brist på enzymer som tillhandahåller energiproduktion, främst på grund av en störning i pyruvinsyrametabolismen och en defekt i elektrontransporten i andningskedjan. En brist på pyruvatdehydrogenaskomplexet (a-E1-subenhet), pyruvatkarboxylas, komplex 1 (NAD-koenzym Q-reduktas) och komplex 4 (cytokromoxidas) i andningskedjan utvecklas.
Det har fastställts att defekter i pyruvatkarboxylas, komplex 1 (NAD-koenzym Q-reduktas) och komplex 4 (cytokromoxidas) i andningskedjan ärvs autosomalt recessivt, medan defekter i pyruvatdehydrogenaskomplexet (a-E1-subenheten) ärvs X-länkat recessivt. Vid punktmutationer i mtDNA, som påverkar den 6:e subenheten av ATPas, är mitokondriell nedärvning typisk. Oftast inträffar en miscens-mutation, associerad med att tymin ersätts med guanin eller cytosin vid position 8993 i mtDNA. Mindre vanligt är en mutation vid position 9176 i mtDNA. På grund av att T8993G-mutationen är den huvudsakliga defekten i NARP-syndrom har familjer med dessa två sjukdomar beskrivits. Hos barn har även en mutation i mtDNA vid position 8344 beskrivits, vilket förekommer vid MERRF-syndrom.
Det antas att vid ansamling av mutant mtDNA i de flesta mitokondrier utvecklas ett allvarligt förlopp av Leighs syndrom. Vid den mitokondriell uppkomsten av detta tillstånd finns mutant mtDNA i 90 % av alla mitokondrier. Patogenesen är förknippad med en kränkning av energibildningen i cellerna och utvecklingen av mjölksyraacidos.
Symtom av Leahs syndrom
De första tecknen på sjukdomen debuterar i tidig ålder (1-3 år). Det finns dock kända fall där sjukdomen manifesterar sig vid 2 veckor och vid 6-7 års ålder. Först utvecklas ospecifika störningar: försenad psykomotorisk utveckling, minskad aptit, kräkningar, viktminskning. Därefter ökar neurologiska symtom: muskelhypotoni eller dystoni med övergång till hypertoni, myokloniska eller tonisk-kloniska anfall, tremor i extremiteterna, koreoatetos, koordinationsstörning, minskade senreflexer, letargi, dåsighet. Cerebral neurodegeneration är progressiv. Symtom på pyramidal och extrapyramidal insufficiens ökar, sväljningsförmågan försämras. Sådana förändringar i synorganet som ptos, oftalmoplegi, atrofi av synnerverna, mer sällan pigmentdegeneration av näthinnan observeras ofta. Ibland utvecklas hypertrofisk kardiomyopati, episoder av takypné uppträder.
I sällsynta fall förlöper sjukdomen som akut encefalopati. Mer typiskt är ett kroniskt eller subakut förlopp, vilket leder till döden flera år efter sjukdomens debut. Vid ett snabbt förlopp (flera veckor) inträffar döden till följd av förlamning av andningscentret.
Diagnostik av Leahs syndrom
Ett biokemiskt blodprov avslöjar laktatacidos på grund av ansamling av mjölksyra och pyrodruvsyra i blodet och cerebrospinalvätskan, samt en ökning av alaninhalten i blodet. Nivån av ketonkroppar kan också vara förhöjd. Ökad utsöndring av organiska syror detekteras i urinen: mjölksyra, fumarsyra etc. Nivån av karnitin i blodet och vävnaderna minskar ofta.
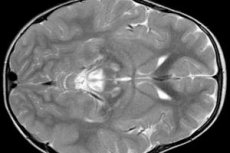
EEG-resultat visar fokala tecken på epileptisk aktivitet. MR-data visar förstoring av hjärnventriklarna, bilateral hjärnskada, förkalkning av basala ganglierna (nucleus caudatus, putamen, substantia nigra, globus pallidus). Atrofi av hjärnhalvorna och hjärnsubstansen kan också detekteras.
Morfologisk undersökning avslöjar grova förändringar i hjärnsubstansen: symmetriska nekrosfokus, demyelinisering och svampig degeneration av hjärnan, främst i mellersta delen, pons, basala ganglier, talamus och synnerv. Den histologiska bilden inkluderar cystisk degeneration av hjärnvävnad, astrocytisk glios, neuronal död och en ökning av antalet mitokondrier i cellerna. I skelettmusklerna finns ansamling av lipidinneslutningar, en minskning av den histokemiska reaktionen på komplex 1 och 4 i andningskedjan, subsarkolemmal ansamling av mitokondrier, onormala mitokondrier med desorganisation av cristae. RRF-fenomenet detekteras ofta inte.
Hur man undersöker?
Vilka tester behövs?
Использованная литература