Medicinsk expert av artikeln

Nya publikationer
Martin-Bells syndrom
Senast recenserade: 04.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
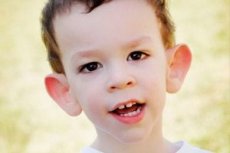
Martin-Bells syndrom beskrevs 1943 av läkare, efter vilka det fick sitt namn. Sjukdomen är en genetisk sjukdom som består av utvecklingsstörning. År 1969 identifierades förändringar i kromosom X (fragilitet i den distala armen) som är karakteristiska för denna sjukdom. År 1991 upptäckte forskare genen som är ansvarig för utvecklingen av denna sjukdom. Denna sjukdom kallas också "fragilt X-syndrom". Både pojkar och flickor är mottagliga för sjukdomen, men pojkar drabbas oftare (3 gånger).
Epidemiologi
Martin-Bell syndrom är en ganska vanlig sjukdom: 0,3-1,0 av 1 000 män lider av denna sjukdom och 0,2-0,6 av 1 000 kvinnor. Dessutom föds barn med Martin-Bell syndrom på alla kontinenter med samma frekvens. Nationalitet, hudfärg, ögonform, levnadsförhållanden och människors välbefinnande påverkar uppenbarligen inte sjukdomens förekomst. Dess förekomstfrekvens är endast jämförbar med frekvensen av Downs syndrom (1 sjukdom av 600-800 nyfödda). En femtedel av de manliga bärarna av den förändrade genen är friska, har inga kliniska eller genetiska avvikelser, resten har tecken på utvecklingsstörning från mild till svår form. Bland kvinnliga bärare är drygt en tredjedel sjuka.
Fragilt X-syndrom drabbar ungefär 1 av 2 500–4 000 män och 1 av 7 000–8 000 kvinnor. Bärarprevalensen bland kvinnor uppskattas till 1 av 130–250; bärarprevalensen bland män uppskattas till 1 av 250–800.
Orsaker Martin-Bells syndrom
Martin-Bell syndrom utvecklas på grund av att kroppens produktion av ett specifikt protein helt eller delvis upphör. Detta sker på grund av bristande respons från FMR1-genen, lokaliserad i X-kromosomen. Mutationen sker som ett resultat av omstruktureringen av genen från instabila strukturella varianter av gentillstånden (alleler), och inte från början. Sjukdomen överförs endast genom den manliga linjen, och mannen behöver inte nödvändigtvis vara sjuk. Manliga bärare överför genen till sina döttrar i oförändrad form, så deras mentala retardation är inte uppenbar. Vid vidare överföring av genen från modern till hennes barn muterar genen, och alla tecken som är karakteristiska för denna sjukdom uppträder.
Patogenes
Patogenesen för Martin-Bell syndrom baseras på mutationer i genapparaten, vilket leder till blockering av produktionen av FMR-protein, ett protein som är livsviktigt för kroppen, särskilt i nervceller, och som finns i olika vävnader. Forskning visar att FMR-proteiner är direkt involverade i processerna för att reglera translationer som sker i hjärnvävnad. Avsaknaden av detta protein eller dess begränsade produktion av kroppen leder till utvecklingsstörning.
I sjukdomens patogenes anses genhypermetylering vara en viktig sjukdom, men det har ännu inte varit möjligt att definitivt identifiera mekanismen för utvecklingen av denna sjukdom.
Samtidigt upptäcktes även locusheterogenitet i patologin, vilket är associerat med polyallelism, såväl som polylokus. Förekomsten av alleliska varianter av sjukdomsutvecklingen fastställdes, vilka orsakas av förekomsten av punktmutationer, såväl som förstörelsen av FMRL-typgenen.
Patienterna har också två fragila tripletter som är känsliga för folsyra, belägna 300 kb, samt 1,5-2 miljoner bp från den fragila tripletten som innehåller FMR1-genen. Mekanismen för mutationer som förekommer i FRAXE- och FRAXF-generna (de identifieras i de ovan nämnda fragila tripletterna) är relaterad till mekanismen för störningar vid Martin Bells syndrom. Denna mekanism orsakas av spridningen av GCC- och CGG-upprepningar, vilket orsakar metylering av de så kallade CpG-öarna. Förutom den klassiska formen av patologin finns det också två sällsynta typer som skiljer sig åt på grund av expansionen av trinukleotidupprepningar (vid manlig och kvinnlig meios).
Det visade sig att patienten vid den klassiska formen av syndromet saknar ett speciellt nukleocytoplasmatiskt protein av typen FMR1, vilket utför funktionen att binda olika mRNA. Dessutom främjar detta protein bildandet av ett komplex som hjälper till att genomföra translationsprocesser inuti ribosomer.
Symtom Martin-Bells syndrom
Hur känner man igen sjukdomen hos barn? Vilka är de första tecknen? Under de första månaderna av ett barns liv är det omöjligt att känna igen Martin-Bell-symtomet, förutom att ibland observeras en minskning av muskeltonus. Efter ett år är den kliniska bilden av sjukdomen mer uppenbar: barnet börjar gå och prata sent, ibland är talet helt frånvarande. Han är hyperaktiv, viftar med armarna slumpmässigt, är rädd för folkmassor och buller, är envis, det finns skarpa vredesutbrott, emotionell instabilitet, epileptiska anfall uppstår, får ingen ögonkontakt. Hos patienter med Martin-Bell syndrom avslöjas sjukdomen också av deras utseende: öronen är utskjutande och stora, pannan är tung, ansiktet är förlängt, hakan är utskjutande, skelning, breda händer och fötter. De kännetecknas också av endokrina störningar: ofta hög vikt, fetma, stora testiklar hos män, tidig pubertet.

Bland patienter med Martin-Bells syndrom varierar intelligensnivån kraftigt: från mild utvecklingsstörning till svåra fall. Om en normal person har en intelligenskvot (IQ) på 100 i genomsnitt, och ett geni har 130, så har personer som är mottagliga för sjukdomen 35-70.
Alla kliniska symtom på patologin kan karakteriseras av en triad av huvudmanifestationer:
- oligofreni (IQ är 35-50);
- dysmorfofobi (utskjutande öron och prognatism observeras);
- makroorkidism, som uppträder efter pubertetens början.
Cirka 80 % av patienterna har också bikuspidalklaffprolaps.
Syndromets fullständiga form manifesterar sig dock endast hos 60 % av alla patienter. Hos 10 % upptäcks endast psykisk retardation, och hos resten utvecklas sjukdomen med en annan kombination av symtom.
Bland de första tecknen på sjukdomen, som uppträder i tidig ålder:
- det sjuka barnet uppvisar betydande utvecklingsstörning i jämförelse med andra jämnårigas utveckling;
- uppmärksamhets- och koncentrationsstörningar;
- stark envishet;
- barn börjar gå och prata ganska sent;
- hyperaktivitet och talutvecklingsstörningar observeras;
- mycket starka och okontrollerbara vredesutbrott;
- mutism kan utvecklas - detta är en fullständig brist på talförmåga hos ett barn;
- barnet upplever social ångest och kan få panik på grund av höga ljud eller andra höga ljud;
- barnet viftar okontrollerat och kaotiskt med armarna;
- blyghet observeras, barnet är rädd för att vara på trånga platser;
- framväxten av olika tvångstankar, instabilt känslomässigt tillstånd;
- Barnet kan vara ovilligt att få ögonkontakt med människor.
Hos vuxna observeras följande symtom på patologin:
- specifikt utseende: ett avlångt ansikte med en tung panna, stora utskjutande öron, en starkt utskjutande haka;
- platta fötter, otit och strabismus;
- puberteten inträffar ganska tidigt;
- fetma kan utvecklas;
- Ganska ofta observeras hjärtfel vid Martin-Bell syndrom;
- hos män observeras förstoring av testiklarna;
- ledernas artikulationer blir mycket rörliga;
- vikt och längd ökar kraftigt.
Diagnostik Martin-Bells syndrom
För att diagnostisera Martin Bells syndrom behöver du kontakta en kvalificerad genetiker. Diagnosen ställs efter specifika genetiska tester som gör att du kan identifiera den defekta kromosomen.
Tester
I ett tidigt skede av sjukdomen används en cytogenetisk metod, där ett fragment av cellmaterial tas från patienten, till vilket folsyra sedan tillsätts för att framkalla förändringar i kromosomerna. Efter en viss tid identifieras ett område av kromosomen där det finns en märkbar förtunning – detta är ett tecken på förekomsten av fragilt X-syndrom.
Detta test är dock inte lämpligt för diagnos i sjukdomens senare skeden, eftersom dess noggrannhet minskar av den utbredda användningen av multivitaminer som innehåller folsyra.
Integrerad diagnostik av Martin-Bells syndrom är en molekylärgenetisk undersökning, som består av att bestämma antalet så kallade trinukleotidupprepningar i genen.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Instrumentell diagnostik
En mycket specifik metod för instrumentell diagnostik är PCR (polymeraskedjereaktion), som gör det möjligt att studera strukturen av aminosyrarester som finns i X-kromosomen och därigenom bestämma förekomsten av Martin Bells syndrom.
Det finns också en separat, ännu mer specifik, metod för patologidiagnostik – en kombination av PCR och detektion med kapillärelektrofores. Denna metod är mycket noggrann och detekterar kromosomal patologi hos patienter med primär ovariell insufficiens, såväl som ataxiskt syndrom.
Förekomsten av defekten kan fastställas efter att ha utfört diagnostik på EEG. Patienter med denna sjukdom har liknande bioelektrisk hjärnaktivitet.
Vilka tester behövs?
Differentiell diagnos
Differentierade metoder som hjälper till att misstänka syndromet inkluderar:
- klinisk - 97,5 % av patienterna har tydliga tecken på utvecklingsstörning (måttlig eller grav); 62 % har utstående stora öron; 68,4 % har en stor utstående haka och panna; 68,4 % av pojkarna har förstorade testiklar, 41,4 % har talsvårigheter (ojämn talhastighet, okontrollerbar volym etc.);
- cytogen - blod undersöks för lymfocytodling, antalet celler med bräcklig X-kromosom per 100 studerade celler bestäms;
- Elektroencefalografi - förändringar i hjärnans elektriska impulser som är specifika för Martin-Bell syndrom registreras.
Vem ska du kontakta?
Behandling Martin-Bells syndrom
Vid behandling av vuxna patienter används antidepressiva medel med psykostimulantia. Läkemedelsbehandlingsprocessen övervakas ständigt av en psykolog och psykiater. Dessutom utförs mikroinjektionsprocedurer med läkemedel som Cerebrolysin (eller dess derivat), samt cytomediner (såsom Solcoseryl eller Lidase) på privata kliniker.
Vid utveckling av ataxiskt syndrom används blodförtunnande läkemedel och nootropika. Dessutom förskrivs aminosyrablandningar och angioprotektorer. Kvinnor med primär äggstockssvikt förskrivs korrigerande behandling med växtbaserade läkemedel och östrogener.
Glutaminreceptorantagonister används också vid behandling.
Traditionellt sett innebär behandlingen av Martin-Bell syndrom användning av läkemedel som påverkar sjukdomens symtom, men inte dess orsak. Denna terapi innebär att man förskriver antidepressiva medel, neuroleptika och psykostimulantia. Inte alla läkemedel är indicerade för användning hos barn, så listan över läkemedel är ganska begränsad. Neuroleptika som kan användas efter 3 år (den tidigaste åldern för deras förskrivning) inkluderar haloperidol i droppar och tabletter, klorpromazin i lösning och periciazin i droppar. Således beräknas dosen av haloperidol för barn beroende på kroppsvikt. För vuxna förskrivs dosen individuellt. Det tas oralt, med början med 0,5–5 mg 2–3 gånger per dag, sedan ökas dosen gradvis till 10–15 mg. När förbättring sker byter man till en lägre dos för att bibehålla det uppnådda tillståndet. Vid psykomotorisk agitation förskrivs 5–10 mg intramuskulärt eller intravenöst, flera upprepningar är möjliga efter 30–40 minuter. Den dagliga dosen bör inte överstiga 100 mg. Biverkningar i form av illamående, kräkningar, muskelspasmer, ökat blodtryck, arytmi etc. är möjliga. Äldre personer bör vidta särskilda försiktighetsåtgärder, eftersom fall av plötsligt hjärtstillestånd har registrerats och tardiv dyskinesi (ofrivilliga rörelser) kan förekomma.
Antidepressiva medel ökar aktiviteten i hjärnstrukturer, lindrar depression, spänningar och förbättrar humöret. Dessa läkemedel, som rekommenderas för användning från 5-8 års ålder vid Martin-Bells syndrom, inkluderar klomipromin, sertralin, fluoxetin och fluvoxamin. Fluoxetin tas därför oralt under måltider 1-2 gånger (helst under första halvan av dagen), med början från 20 mg per dag, ökande till 80 mg vid behov. Äldre personer rekommenderas inte en högre dos än 60 mg. Behandlingsförloppet bestäms av en läkare, men inte mer än 5 veckor.
Möjliga biverkningar: yrsel, ångest, tinnitus, aptitlöshet, takykardi, ödem etc. Försiktighet krävs vid förskrivning till äldre, personer med hjärt-kärlsjukdomar och diabetes.
Psykostimulantia är psykotropa läkemedel som används för att förbättra uppfattningen av yttre stimuli: de skärper hörsel, reaktioner och syn.
Diazepam förskrivs som ett lugnande medel för neuroser, ångest, epileptiska anfall och kramper. Det tas oralt, intravenöst, intramuskulärt, rektalt (i ändtarmen). Det förskrivs individuellt, beroende på sjukdomens svårighetsgrad, med de lägsta doserna på 5-10 mg, dagligen - 5-20 mg. Behandlingstiden är 2-3 månader. För barn beräknas dosen med hänsyn till kroppsvikt och individuella egenskaper. Biverkningar inkluderar letargi, apati, dåsighet, illamående, förstoppning. Det är farligt att kombinera med alkohol, beroende av läkemedlet är möjligt.
Vid behandling av Martin-Bells syndrom har det förekommit fall av förbättring av tillståndet och med införandet av läkemedel tillverkade av animaliskt material (hjärnan): cerebrolysat, cerebrolysin, cerebrolysat-M. Huvudkomponenterna i dessa läkemedel är peptider som främjar proteinproduktionen i neuroner och därmed fyller på det saknade proteinet. Cerebrolysin administreras som en stråle på 5-10 ml, behandlingsförloppet består av 20-30 injektioner. Läkemedlet förskrivs till barn från ett års ålder, administreras intramuskulärt varje dag 1-2 ml i en månad. Upprepade administreringssessioner är möjliga. Biverkningar i form av feber, kontraindicerade för gravida kvinnor.
Det gjordes försök att behandla sjukdomen med folsyra, men endast beteendeaspekten förbättrades (nivån av aggression och hyperaktivitet minskade, talet förbättrades), och ingenting förändrades på den intellektuella nivån. För att förbättra sjukdomstillståndet ordineras folsyra, fysioterapimetoder, logopedi, pedagogisk och social korrigering indikeras.
Litiumpreparat anses också vara effektiva, eftersom de bidrar till att förbättra patientens anpassning till den sociala miljön, såväl som kognitiv aktivitet. Dessutom reglerar de även dennes beteende i samhället.
Örter kan användas vid Martin-Bell syndrom som antidepressiva medel. Örter som hjälper till att lindra spänningar, ångest och förbättra sömnen inkluderar valeriana, pepparmynta, timjan, johannesört och kamomill. Infusioner bereds enligt följande: för 1 tesked torra örter behöver du ett glas kokande vatten, avkok dras i minst 20 minuter, tas huvudsakligen på kvällen före sänggåendet eller på eftermiddagen. En sked honung skulle vara ett bra komplement till dem.
Sjukgymnastikbehandling
För att eliminera neurologiska manifestationer utförs speciella fysioterapeutiska procedurer - såsom övningar i poolen, muskelavslappning och akupunktur.
Kirurgisk behandling
Ett viktigt behandlingssteg anses också vara plastikkirurgiska metoder - operationer som hjälper till att förbättra patientens utseende. Plastikkirurgi utförs på lemmar och öron, såväl som på könsorganen. Korrigering av gynekomasti med epispadi utförs också, liksom andra defekter i utseendet.
Förebyggande
Den enda metoden för att förebygga sjukdomen är prenatal screening av gravida kvinnor. Det finns speciella undersökningar som möjliggör tidig upptäckt av patologin, varefter det rekommenderas att avbryta graviditeten. Som ett alternativ används IVF, vilket kan hjälpa barnet att ärva en frisk X-kromosom.
Förebyggande av patientens sjukdom beror på om genmutationen har uppstått igen eller ärvts. För detta utförs molekylärgenetisk diagnostik. Det faktum att testet inte avslöjade den "bräckliga X-kromosomen" hos släktingar talar för mutationens "färska", vilket innebär att risken att få ett barn med Martin-Bells syndrom är mycket liten. I familjer där det finns sjuka personer kommer testet att bidra till att undvika upprepade fall.
Prognos
Prognosen för Martin-Bells syndrom är gynnsam för livet, men inte för återhämtning. Livslängden beror på sjukdomens svårighetsgrad och associerade defekter. Patienten kan leva ett normalt liv. Vid svåra former av Martin-Bells syndrom riskerar patienter att drabbas av livslång funktionsnedsättning.
Förväntad livslängd
Martin Bells syndrom har ingen allvarlig negativ inverkan på hälsan, så livslängden för de flesta som har diagnostiserats med denna patologi skiljer sig inte från standardindikatorer.