Medicinsk expert av artikeln

Nya publikationer
Angelmann syndrom hos barn och vuxna
Senast recenserade: 23.04.2024

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.

Det finns ett antal sjukdomar där uttryck som "ta hand om dig själv och inte bli sjuk" ljud, åtminstone löjligt. Denna patologi, där vissa psykiska och fysiska avvikelser är inbäddade i ett barns kropp redan före födseln, men föräldrarna har ingen skuld. Sådana sjukdomar orsakas av mutationer eller störningar i kromosomsatser och kallas kromosomala eller genetiska. Angelman syndrom, Downs syndrom, Patau, Edwards, Turner, Prader-Willi är bara en del av de genetiska sjukdomarna från en ganska anständig lista.
Syndrom av en glad person
Den här gången vi talar om sjukdomen, uppkallad efter den brittiska barnläkaren Harry Angelman från det första tog upp frågan om problemet 1965 år, inför på tröskeln till sin praktik med tre ovanliga barn, förenas av gemensamma säregna symtom. Läkaren heter dessa barn marionettbarn och skrev om dem en artikel som ursprungligen kallades "marionettbarn". Artikeln själv och dess namn skrevs under intrycket av en bild som ses på ett av museerna i Verona. Bilden avbildade en skrattande pojke, och den kallades "Boy-marionett". Föreningen bilden av barnet med de tre barnen med Angelmans som en gång ställs inför i sin praktik, och sköt barnläkare barn kombinera till en grupp på grund av deras befintligt skick.
Det faktum att barnen noterade i artikeln inte märktes av andra läkare är inte överraskande. När allt började verkade det vid första anblicken att de hade helt olika sjukdomar, så den allmänna kliniska bilden av sjukdomen skilde sig i tre olika fall. En "ny" kromosomal patologi kan vara intressant för andra forskare, men den tiden var genetiken ännu inte utvecklad för att bekräfta hypotesen för en engelsk läkare. Därför var artikeln efter ett visst intresse för det länge övergiven till det avlägsna regementet.
Nästa omnämnande av Angelman syndromet, och så kallades det nu artikeln från barnläkaren från England, G. Anglemann, från början av 80-talet av 20-talet. Och först år 1987 var det möjligt att hitta orsaken till att en liten del av barnen föddes med sådana avvikelser, att från sidan verkar de ständigt leende och glada. Det är faktiskt inte så, och leendet är bara en grimas, bakom vilken ligger den olyckliga människans själ och föräldrarnas smärta.
Epidemiologi
Kromosomalmutation i ett barn, enligt statistik, kan utvecklas både mot bakgrunden av sådana mutationer i föräldrarna och i avsaknad av sådana. Det finns ingen klar ärftlig karaktär i Angelman syndromet (SA), men sannolikheten för att utveckla patologi hos föräldrar med kromosomala mutationer är ganska hög.
Det är också intressant att om en familj redan har ett barn med en SA, finns det en procent risk att ha ett andra barn av samma typ, även om föräldrarna är hälsosamma.
Det finns fortfarande ingen exakt statistik om antalet patienter med Anghelmans syndrom. Kanske är felet ett antal symptom som kan uppstå i en viss komposition eller under lång tid uppstår inte alls. Det antas att förekomsten av sjukdomen är: 1 barn per 20.000 nyfödda. Men den här siffran är mycket ungefärlig.
Orsaker ängelman syndrom
Angelmann syndrom är det medicinska namnet för kromosomal patologi, men det är inte den enda. Hos människor kallas denna sjukdom också marionettbarns syndrom, syndromet hos en lycklig marionett, Petrushka syndromet och en skrattdockas syndrom. Ja vilken typ av namn kan människor inte komma med (ibland till och med förolämpande för patienterna själva och deras föräldrar), men sjukdomen är en sjukdom, oavsett hur roligt det ser utåt och vad som helst orsakerna kan orsakas.
Och orsakerna till utvecklingen av Angelman-syndromet, liksom många andra genetiska patologier, är i alla fall kränkningar i strukturen hos en av kromosomerna eller kromosomen som helhet. Men bara i vårt fall ligger hela problemet i de 15 kromosomerna som överförs från moderen. Dvs Faderns kromosom har i detta fall inga avvikelser, men kvinnan genomgår vissa mutationer.
Enligt typen av kromosomal abnormitet refererar Angelmanns syndrom till kromosomala mutationer. Sådana mutationer är:
- Deletion (avsaknad av kromosomregion som innehåller en specifik uppsättning av gener, och om ingen av generna det kommer microdeletions), vilket är ett resultat av två diskontinuiteter och en återförening när förlorade del av den ursprungliga kromosomen.
- Dubblering (närvaron av en extra plats i kromosomen, som är en kopia av den redan tillgängliga), som i de flesta fall leder till en persons död, mindre ofta - till infertilitet.
- Inversion (inversion av en kromosom sektioner vid 180 grader, dvs i motsatt riktning, och sedan gener däri är anordnade i omvänd ordning) när den brutna kromosomen ändar anslutna i en ordning som skiljer sig från den ursprungliga.
- Insertion (om en del av det genetiska materialet i kromosomen inte finns på plats)
- translokation (om en del av kromosomen sammanfogar en annan kromosom, kan en sådan mutation vara ömsesidig utan förlust av platser).
Att få en muterad kromosom från en intet ont anande mamma, är barnet dömt att födas med avvikelser i förväg. Den vanligaste orsaken till Anghelmans syndromutveckling är fortfarande delningen av mammans 15 kromosom, när det inte finns något litet område i den. Mindre vanliga mutationer i syndromet i "skratta dockan" är:
- translokation,
- enfaddiosomi (om barnet fick ett par kromosomer från fadern, är moderkromosomen frånvarande)
- mutation av gener i DNA, som är både huvudbyggnadsmaterialet (genetiskt) material och instruktionen för dess korrekta användning (i synnerhet mutationen av ube3a-genen i moderkromosomen).
Förekomsten av en sådan mutation hos föräldrarna är en riskfaktor för Anghelman syndrom hos barn. Men inte bara kromosomala mutationer, men genomet (som är associerade med en kvantitativ förändring i kromosomuppsättningar och mer vanligt kromosom) kan framkalla utveckling av sjukdom hos barnet. Till vanliga genomiska mutationer kan tillskrivas tromomi av kromosomer (om en person har en kromosomsats har mer än 46 kromosomer).
Till patologin hos barnet behöver inte nödvändigtvis ha föräldrar kromosomala abnormiteter. Och ändå finns det en viss procent av patienter vars sjukdom är ärftlig.

Patogenes
Låt oss gräva lite i biologi, mer exakt i genetiken. Den genetiska informationen för varje enskild mänsklig kropp finns i 23 par kromosomer. En kromosom från paret överförs till barnet från fadern, den andra från moderen. Alla par av kromosomer skiljer sig i form och storlek och bär i sig viss information. Så är 23 par kromosomer (X och Y kromosomer) ansvariga för bildandet av barnets sexuella egenskaper (XX - en tjej, XY-pojke, medan ett barn kan få en Y-kromosom från sin far bara).
Helst får ett barn från sina föräldrar 46 kromosomer, som bildar sina genetiska egenskaper, bestämmer honom som individ. Ett större antal kromosomer kallas trisomi och anses vara en avvikelse från normen. Till exempel orsakar närvaron av 47 kromosomer i kromosomsatsen (karyotyp, som bestämmer art och individuella egenskaper) uppkomsten av Downs syndrom.
Om kromosomerna är tonade med ett speciellt färgämne, så kan man i mikroskopet se band av olika nyanser längs var och en av dem. Inom varje band finns ett stort antal gener. Alla dessa band är numrerade av forskare och har en fast plats. Frånvaron av ett av banden anses vara en avvikelse från normen. När Angelmans syndrom kan ofta observera frånvaron av maternkromosomsegment mellan Q11-Q13, är belägna i den långa armen, antalet DNA-baser som endast ca 4 miljoner.
Kromosomens huvudkomponent är en otroligt lång DNA-molekyl som innehåller tusentals gener och tiotals hundratals miljoner kvävebaser. Således innehåller de 15 kromosomer som är ansvariga för utvecklingen av Angelmans syndrom och flera andra 1200 ar och cirka 100 miljoner baser. Eventuella överträdelser i DNA-molekylens struktur påverkar nödvändigtvis utseendet och utvecklingen av det ofödda barnet.
Den genetiska informationen som finns i generna omvandlas till ett protein eller RNA. Denna process kallas genuttryck. Således får den genetiska informationen från föräldrarna både form och innehåll, som är förkroppsligade i deras unika arvtagare till kvinnligt eller manligt kön.
Det finns ett antal sjukdomar med nonclassical typ av arv, inklusive Angelmans syndrom, där generna fått från föräldrar som en del av de parade kromosomer unika avtryck föräldrar och yttrar sig på olika sätt.
Så, är Angelmans syndrom ett utmärkt exempel på den genetiska präglingen, varvid uttrycket av gener i kroppen hos barnet är direkt beroende på från vilken moder härledda alleler (olika former av samma gen erhållen från fadern och modern är belägna på de identiska partierna av parade kromosomer) . Dvs till framväxten av syndromet leder endast anomalier i moderkromosomen, medan mutationer och störningar i strukturen hos den faderliga kromosomen orsakar helt olika patologier.
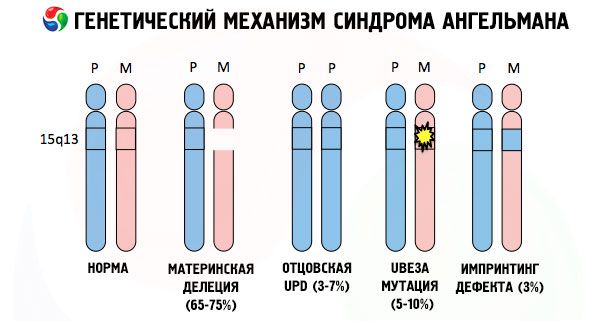
I denna sjukdom finns det en brist av specifika gener i moderns kromosom eller förlusten / minskning i aktiviteten av enskilda gener (i de flesta fall ube3a gen inblandad i metabolismen Ubiquitin - proteinnedbrytning andra regulatoriska proteiner). Som ett resultat av detta diagnostiseras barnet med psykiska utvecklingsavvikelser och fysiska missbildningar.
Symtom ängelman syndrom
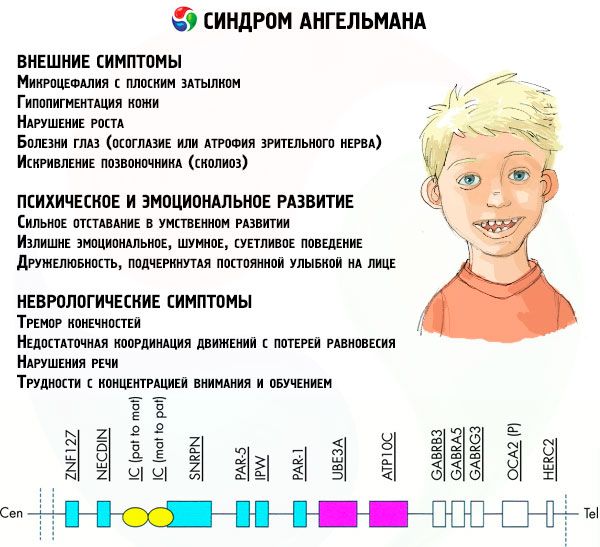
Symptomen på Angelmans syndrom påverkar olika aspekter av ett barns liv och utveckling: fysisk, neurologisk och psykisk. Baserat på detta kan vi skilja 3 grupper av symtom som indikerar utvecklingen av denna patologi.
- Externa eller fysiska symptom:
- ett oproportionerligt litet huvud jämfört med stammen och lemmarna som är av normal storlek,
- för bred mun,
- på ansiktet finns nästan alltid ett leende (med öppen mun)
- sällsynta tänder,
- smal överläpp,
- sticker ofta ut en bred tunga,
- utskjutande nedre käften,
- skarp haka,
- mycket lätt hud, ofta också hår (albinism, förknippad med det faktum att kroppen inte producerar pigmentmelanin),
- mörka fläckar på lätt hud (hypopigmentering på grund av otillräcklig produktion av melanin)
- fysiska eller yttre symtom: ögonsjukdomar, såsom strålning eller atrofi hos optisk nerv,
- korsning av ryggraden (skolios)
- styva ben (när man går på en man böjer inte knäna på grund av den lilla rörligheten i lederna, därav jämförelsen med marionetten).
- Symtom i samband med mental och emotionell utveckling:
- en stark fördröjning i mental utveckling,
- onödigt emotionellt, bullrigt, noga beteende,
- frekvent klappning,
- den uttryckta vänligheten, betonad av ett konstant leende på ansiktet,
- täta, orsakslösa skratt.
- Neurologiska symptom:
- tremor av extremiteter,
- otillräcklig samordning av rörelser med förlust av balans,
- minskad muskelton,
- en mängd olika sömnstörningar,
- frekventa hysteriska anfall i barndomen,
- talförlust (barnet börjar prata sent, har dåliga kommunikationsförmåga och slumrat tal),
- hyperaktivitet mot bakgrund av ökad excitabilitet,
- svårigheter med koncentration och träning.
Men det här är en generaliserad bild av sjukdomen. Faktum är att den kliniska bilden av Anghelmans syndrom i stor utsträckning beror på sjukdomsutvecklingsstadiet och typen av kromosommutation som orsakade patologin. Och detta betyder att hos olika patienter kan symtomatologin av sjukdomen skilja sig avsevärt, vilket under lång tid inte möjliggjorde isolering av patologi bland annat med en liknande klinisk bild.
Bland det totala antalet symtom kan identifieras som är karakteristiska för alla patienter utan undantag:
- svåra avvikelser i mental utveckling,
- otillräckligt beteende (oavsiktligt skratt, ökad excitabilitet, dålig koncentration av uppmärksamhet, ett tillstånd av eufori),
- underutveckling av motoriska färdigheter,
- dålig samordning av rörelser, walking ataxi (ojämn takt, svängande från sida till sida, etc.), tremor av extremiteter.
- brott mot talutveckling med övervägande av icke-verbala kommunikationsmedel.
Bland de symptom som uppträder hos de allra flesta patienter kan vi skilja på följande:
- oproportionerligt huvud och bagage, orsakad av en försening i fysisk utveckling,
- hos många patienter är formen på skallen sådan att hjärnans storlek är mindre än hos friska människor (mikrocefali),
- epileptiska anfall i en ålder av upp till 3 år med en progressiv minskning av styrka och frekvens hos äldre,
- förvrängning av EEG-index (oscillationer och hög amplitud av lågfrekventa vågor).
Dessa symtom uppträder ganska ofta, men i 20% av patienterna med Angelmanns syndrom är de frånvarande.
Ännu mer sällan kan diagnostisera sådana manifestationer av sjukdomen som:
- uttalad eller liten strabismus,
- en svag kontroll över tungans rörelse, vilket leder till att patienter ofta sticker ut tungan utan anledning,
- svårigheter att svälja och suga, särskilt hos yngre barn,
- brott mot hud- och ögonpigmentering,
- upphöjda eller böjda under gånghänder,
- giperrefleksiya,
- sömnstörningar, särskilt i barndomen,
- frekvent salivation,
- irrepressibel törst,
- överdriven aktiva tuggrörelser,
- överkänslighet mot värme,
- platt huvud,
- avancerad underkäke,
- släta palmer.
En ganska stor andel av patienterna har problem med urinering, vilket de dåligt kontrollerar, kränkning av bra motoriska färdigheter, vilket skapar svårigheter med självbetjäning och träning, övervikt. Praktiskt taget i alla patienter börjar puberteten senare än hos friska kollegor.
Barn med ängelman syndrom är bra på att förstå och förstå muntligt tal, men vill inte delta i konversationen, vilket begränsar sitt tal till dussintals ord som behövs i vardagen. Men i vuxen ålder ser dessa patienter yngre ut än sina kamrater utan genetiska patologier.
Många av symtomen på Angelmanns syndrom är svaga, så den kliniska bilden av sjukdomen förändras med ålder. Kramper och epileptiska anfall blir mer sällsynta eller försvinner alls, patienten blir mindre agiterad, sömnuppsättningar.
Komplikationer och konsekvenser
Angelman syndrom är en allvarlig, praktiskt taget obotlig kromosomal patologi, som berövar patienter möjligheten att leva ett normalt liv. Vad blir barnets liv med SA, beror i stor utsträckning på typen av kromosomal abnormitet.
Dubblering av kromosomregionen är i de flesta fall oförenlig med livet. Och även om sådana patienter inte dör i spädbarn och når puberteten, har de ingen möjlighet att få barn.
Deletion eller frånvaro av en del av de gener som oftast förekommer i Anghelmans syndrom är ett hinder för att barnet lär sig att gå och prata. Sådana barn har utvecklingsstörning förekommer i allvarligare, ofta haft ett epileptiskt anfall och deras intensitet är mycket starkare än för patienter med andra kromosomavvikelser.
Om det bara finns en mutation av en gen, med vederbörlig uppmärksamhet och tillvägagångssätt kan barnet lära sig grunderna om självbetjäning, kommunikation och kommunikation i laget, även om han fortfarande kommer att ligga kvar i utvecklingen från sina kamrater.
För barn med Angelmanns syndrom, välvilligt av naturen, är huvudet kärleken och uppmärksamheten hos föräldrarna. Endast i detta fall kommer barnets träning att bära frukt, även små. Självklart kan patienter i grundskolan inte studera med SA. De behöver specialklasser, där barn först lär sig att koncentrera sin uppmärksamhet, och sedan kommer de gradvis att ge grunderna i skolkunskap.
Diagnostik ängelman syndrom
Angelman syndrom är en medfödd patologi av utveckling. Men på grund av vissa omständigheter är det oftast inte möjligt att diagnostisera det i spädbarn och tidig barndom. Anledningen till detta är nonspecificitet och milda symptom hos spädbarn och småbarn på upp till 3 år. Och förekomsten av sjukdomen i vårt land är inte så stor att läkare har lärt sig att känna igen det bland dess liknande.
Angelmans syndrom hos spädbarn kan visa sig i form av minskad muskeltonus, vilket manifesteras i form av matningsproblem (svaghet sugande och sväljreflex), och senare inlärningssvårigheter till promenader (sådana barn mycket senare börja gå). Dessa symptom är de första tecknen på en avvikelse i utvecklingen av barnet, vilket kan vara väl associerat med en kromosomal abnormitet. Bekräfta detta antagande kan bara genetisk analys.
Särskild uppmärksamhet ägnas åt barn vars föräldrar har olika genomiska eller kromosomala abnormiteter. Efter popervosti sjukdomen inte kan visa sig på något sätt, och om du upptäcker avvikelser i tid, började att kraftfullt engagera sig med barnet, är det möjligt att uppnå mycket större framgång i utbildning, stal utvecklingen av sjukdomen.
Om föräldrarna har olika kromosomala abnormiteter utförs den genetiska analysen redan innan barnet är födt, eftersom CA är en av de patologier som kan detekteras i ett embryonalt tillstånd.
Samlingen av material för genetisk forskning kan göras på två sätt:
- invasiv (med viss riskandel, eftersom det är nödvändigt att gå in i livmodern för att kunna göra ett test av fostervätska)
- icke-invasiv (DNA-analys av barnet av moderns blod).
Sedan utförs följande undersökningar:
- fluorescerande in situ-hybridisering (FISH-metod) -bindning av en DNA-prob märkt med ett speciellt färgämne till det DNA som studeras, följt av en mikroskopisk undersökning.
- analys av mutationer i ube3a-genen och tryckande gener,
- analys av DNA-metylering med hjälp av speciella metoder som används inom genetik.
Genetiska analyser ger rätt noggrann information vid kromosomala abnormiteter, så framtida föräldrar vet i förväg vad de ska vara förberedda för. Ändå finns det undantag. I en viss grupp patienter, i närvaro av alla symptomindikerande symtom, förblir analysresultaten fortfarande normala. Dvs För att avslöja en patologi är det möjligt att endast observera barnet från den tidigaste barndomen: hur äter, när han började gå och prata, om benen böjer när man går etc.
Förutom FISH-metoden, bland metoder för diagnostiskt verktyg Angelmans syndrom kan urskiljas tomografi (CT eller MRI), hjälper till att bestämma status och hjärnstorlek, och elektroencefalogram (EEG), som visar hur de enskilda delarna av hjärnan fungerar.
Läkarnas slutliga diagnos är vanligtvis inställd i åldern 3-7 år, då patienten redan har huvuddelen av symtomen och dynamiken i sjukdomsutvecklingen är synlig.
Vilka tester behövs?
Differentiell diagnos
Angelman syndromet är en genetisk patologi som inte faktiskt har specifika manifestationer. De flesta symtomen kan också indikera både CA och andra genetiska patologier.
Differentiell diagnos i Anghelmans syndrom utförs med följande patologier:
- Pitt-Hopkins syndrom (patienter kännetecknas av mental retardation, glatt karaktär, leende, de har en ganska stor och bred mun, mikrocefali noteras). Skillnad - attacker av hyperventilation och fördröjning av andning i vakna tillstånd.
- Christi syndrom (patienter är utvecklingsstörda människor med en glad disposition, som inte kan tala, de kännetecknas av mikrocefali, ataxi, kramper, ofrivilliga muskelrörelser).
- Mowata-Wilson syndrom (symtom: mental retardation, epileptiska anfall, skarp hakan, öppen mun, uttryck av lycka på ansikte, mikrocefali). Skillnaden är ett stort avstånd mellan ögonen, ögonen är avfasade inåt, näsens spets är avrundad, öronen sitter tillbaka.
- Kabuki syndrom (som kännetecknas av mild till måttlig utvecklingsstörning, problem med tal- och motorik, muskelsvaghet, kramper, mikrocefali, stora spalter mellan zudami, koordinationssvårigheter). Skillnaden - i form av en båge ögonbryn, inverterad laterala delen av det nedre ögonlocket, bred liggande ögon, långa ögon slitsar med långa tjocka fransar.
- Rett syndrom (differentiering med CA hos kvinnor). Symtom: Fördröjd talutveckling, konvulsiva anfall, mikrocefali. Skillnaden - det finns inget lyckligt uttryck på ansiktet, det finns attacker av apné och apraxi, som så småningom fortskrider.
- Syndrom autosomal recessiv mental tardatsii 38 (symptom: mental retardation med en märkbar fördröjning i utvecklingen av motorik och tal, muskelsvaghet, matningsproblem i spädbarnsåldern, impulsivitet). Skillnaden är irisens blå färg.
- Syndrom för duplicering av genen MESR 2 (differentiering med SA hos män). Symtom: allvarlig mental retardation, muskelsvaghet från barndomen, problem med tal eller brist på det, epilepsi. Skillnader - progressiv myopati, ständigt återkommande infektioner.
- Clifstra syndrom (symptomatisk: tal och tänkande problem, muskelsvaghet, sömnstörningar, brist på uppmärksamhet, en lätt öppen mun, hyperaktivitet, krampanfall, ataxi, obalans). Skillnader - ett platt ansikte, kort snubben, breda ögon, stor inverterad underläpp, attacker av aggression.
- Syndrom Smith-Magenis (kännetecknat av anfall, sömnstörningar, störningar i intellektuell och motorisk utveckling). Skillnader - ett brett och platt ansikte, konvex panna.
- Kulena-de Vries syndrom (mild och måttlig mental retardation, muskelsvaghet, krampanfall, vänlighet). Skillnader - ett långt ansikte med hög panna, utstående öron, sneda ögon, ökad rörlighet i lederna, medfödda hjärtpatologier.
- Syndrome Philan - McDermid (symtom: mental retardation, speech impairment eller brist på det). Skillnader - stora händer med utvecklade muskler, muskelsvaghet sedan födseln, svag svettning.
Angelmans syndrom liknande symptom kan "skryta" och hur en sådan patologi adenilsuktsinazy brist, en autosomal recessiv syndrom mental retardation 1 kromosomen 2q23.1 dubbel syndrom, haploinsufficiency gener FOXG1, STXBP1 eller MEF2C och andras.
Doktorns uppgift är att göra en exakt diagnos som skiljer Angelmann syndrom från patologier med liknande symtom och förskriver en effektiv behandling som är relevant för den diagnostiserade graden av sjukdomsutveckling.
Vem ska du kontakta?
Behandling ängelman syndrom
Angelman syndrom avser kategorin av dessa patologier, sökandet efter effektiv behandling av vilken medicin som är förlovad till denna dag. Den etiologiska behandlingen av sjukdomen ligger i utvecklingsstadiet av olika metoder och medel, av vilka många ännu inte har testats hos människor. Hittills läkare måste begränsas till den symptomatiska terapi för att hjälpa på något sätt lindra den svåra situationen för barn och vuxna med marionett syndrom, lider av epileptiska anfall, salivavsöndring, hypotension och sömnstörningar.
Så minska frekvensen och styrkan hos epileptiska anfall kan vara med ett väl valda antikonvulsiva läkemedel. Men hela svårigheten är att anfall hos patienter med AS skiljer sig från de vanliga epileptiska anfallen, eftersom de kännetecknas av flera typer av kramper, vilket innebär att det blir möjligt att lindra tillståndet genom administrering av flera läkemedel samtidigt.
De mest populära antikonvulsiva läkemedel som används för behandling av Anghelman syndrom är: valproinsyra, topiramat, lamotrigin, levetiracetam, klonazepam och preparat baserade på dem. Mindre vanliga droger baserade på karmazepina, fenytoin, fenobarbital, etosuximid, eftersom vissa av dem kan prova en paradoxal effekt, vilket är att öka och öka frekvensen av epileptiska anfall. Detta händer om läkemedlet används som en del av monoterapi.
För behandling av salivation används vanligtvis två metoder: läkemedel (preparat som undertrycker salivbildning) och operationell, bestående av reimplantation av spyttkanalerna. Men i fråga om CA anses dessa metoder vara ineffektiva, och frågan är öppen. Föräldrar och de som bryr sig om sådana patienter, vi måste ägna särskild uppmärksamhet åt detta ögonblick, eftersom patienterna inte brukar kontrollera salivation, och vissa kan helt enkelt inte ta hand om sig själva.
Ett annat problem är den korta varaktigheten av sömnen. Ofta sover barn med Angelmans syndrom inte mer än 5 timmar, vilket negativt påverkar hela organismens arbete. Spännande, aktiva barn, kärleksfulla spel och kommunikation (även om de försöker begränsa sig till icke-verbala sätt), är märkbart trötta för dagen. För att ha en bra vila behöver kroppen en god full sömn, men det är bara problemet med det.
Det verkar för att förbättra sömnen i retbara patienter bör vara tillräckligt läkemedel med lugnande effekter (fentiaziner och atypiska antipsykotika), lugnar nervsystemet. Men i fallet med CA är användningen av sådana droger fyllda med utseendet av negativa effekter. Därför läkare föredrar fortfarande lätt hypnotiska läkemedel, såsom "Melatonin" (naturliga hormonella preparatet på grundval av sömnhormonet), som ger patienterna en timme före sänggåendet på en tablett, och "Diphenhydramine". Frekvensen för administrering och dosering av denna ställs av läkaren beroende på patientens tillstånd och ålder.
Ibland har patienter med ängelman syndrom problem med matsmältning och pall. För att justera en stol är det möjligt med laxerande preparat (det är bättre än en fytogenes).
Och du kan närma sig problemet på olika sätt, liksom de amerikanska läkare, baserat på några av de metoder för behandling av autism, eftersom många av symptomen är karakteristiska för SA, är också utmärkande för autism (impulsivitet, ofrivilliga rörelser, repetitiva handlingar, uppmärksamhet underskott, problem i kommunikationen, etc. ) .. Det har observerats att administrering av hormonet sekretin, normalisera matsmältningen och en stol, en positiv effekt på patienternas uppmärksamhet och oxytocin bidrar till att förbättra barns kognitiva förmågor och minne, för att korrigera beteende.
Det är sant att vissa hormoner är oumbärliga här, särskilt när det gäller barn. Angelman syndrom visar beteendeterapi, arbetar med en psykolog och talterapeut (undervisning i icke-verbala kommunikations- och teckenspråk). Utbildning av sådana barn bör grundas på ett individuellt program med deltagande av specialutbildade lärare, en psykolog och föräldrar. Tyvärr är det inte möjligt överallt, och familjerna är ensamma med sitt problem.
Eftersom många små patienter med CA lider av låga muskeltoner och gemensamma problem uppmärksammas mycket på fysioterapeutisk behandling. Oftast går läkare till användning av paraffinapplikationer, elektroferos, magnetoterapi.
Aktiv toning massage och speciella övningar av sjukgymnastik hjälper det sjuka barnet efter en tid med självförtroende att stå på fötterna och gå. Särskilt användbart i detta avseende aquagymnastik, som rekommenderas i CA i kallt vatten. Det ökar muskeltonen och lär barnet att äga sin kropp, koordinera rörelser.
Antikonvulsiv behandling
Det farligaste symptomet i Anghelmans syndrom är anfall som liknar epileptiska anfall. Detta symptom observeras hos 80% av patienterna, vilket innebär att alla ska ordineras effektiv antikonvulsiv behandling.
Behandling av epileptiska anfall utförs med hjälp av vitaminer och antikonvulsiva medel. När Angelmans syndrom, tillsammans med krampaktig syndrom, kommer att vara användbart vitaminer B-gruppen, liksom vitamin C, D och E. Men vitamin terapi för att utse sina egna i det här fallet är mycket farligt, eftersom okontrollerade intag av vitaminer kan minska effekten av antiepileptiska läkemedel och provocera nya, svår och utdragen attacker.
Valet av antikonvulsiva läkemedel och utnämningen av deras effektiva dosering bör också hanteras av en specialistläkare. Han bestämmer också om det finns tillräckligt med ett läkemedel eller patienten måste ta 2 eller flera läkemedel under lång tid .
De flesta patienter läkare ordinera läkemedel valproinsyra ( "valproinsyra", "Depakinum", "Konvuleks", "valparin" et al.), Som förhindrar kramper, förbättra humöret och psykiska tillståndet hos patienterna.
Valproinsyra finns i form av tabletter, sirap och injicerbara lösningar. Det mest populära läkemedlet är läkemedlet för långvarig åtgärd "Depakin" i tabletter och som lösning för intravenös administrering. Doseringen av läkemedlet bestäms av läkaren individuellt beroende på patientens vikt, ålder och tillstånd.
Ta drogen under måltider 2 till 3 gånger om dagen. Den genomsnittliga dagliga dosen är 20-30 mg per 1 kg av patientens vikt, maximalt 50 mg / kg per dag.
Kontra till användning. Det används inte för brott mot lever och bukspottkörtel, hemorragisk diatese, hepatit, porfyri och överkänslighet mot läkemedlet.
Bland biverkningarna kan man skilja handskakningar, matsmältning och pall, förändringar i kroppsvikt.
"Topiramat" är också ett läkemedel av val i CA. Det är tillverkat i form av tabletter och används som en del av monoterapi och i kombination med andra läkemedel.
Metod för applicering och dosering. Ta piller inuti utan hänsyn till matintag. Det första dagliga intaget för vuxna är 25-50 mg, för barn 0,5-1 mg / kg. Varje vecka ökar dosen enligt doktors recept.
Läkemedlet ska inte tas under graviditet och laktation, liksom med ökad känslighet för dess komponenter. Läkemedlet har många olika biverkningar.
Läkemedel som läkaren kan ordinera vid Angelmans syndrom "Klomazepam", "Rivotril" "Lamotrigin", "Seyzar", "Lamictal", "levetiracetam", "Keppra", "Epiterra" et al.
Alternativ behandling och homeopati
Alternativ medicin, som homeopatiska läkemedel, skiljer sig visserligen från jämförande säkerhet, men här kan effekten av sådan behandling i förhållande till Angelholm syndrom anses vara kontroversiell.
Även om vissa alternativa behandlingar fortfarande kan hjälpa. Det handlar om att stoppa epileptiska anfall. I detta avseende kan örtbehandling vara ganska effektiv.
En bra effekt ges av en medicinsk avgift baserad på peon, lakrits och ankor (komponenter tas i lika stora mängder). Gräs måste slipas i mjöl. Efter 2 veckor från mottagningens början kan du märka en signifikant minskning av frekvensen av konvulsiva attacker.
Användbar för kramper och ett avkok av lavendel (1 tsk för ett glas kokande vatten). Formuleringen kokas i 5 minuter och insisteras i en halvtimme. Ta medicinen över en natt i 14 dagar.
Effektivt för epileptiska attacker betraktas som vatten (eller alkohol) infusion motherwort.
Från homeopati läkemedel för att förhindra anfall med Angelmans syndrom kan användas läkemedel baserade på kamomill och Hjärt, Acidum hydrocyanicum, Argentum NITRICUM, Kalium bromatum, Arsenicum album. Men det är nödvändigt att överväga att effektiva och säkra doser av preparat i varje konkret fall kan utse endast läkaren en homeopatist.
Förebyggande
Som läsaren troligen redan har förstått, för att förhindra mutationen av gener och andra kromosomala avvikelser, är läkemedlet fortfarande bortom makt, liksom att åtgärda situationen. Detta kan hända med alla, eftersom barn med Angelmann syndrom förekommer även i friska föräldrar, och genetik, som för närvarande är en av de minst studerade grenarna av medicin, kan inte förklara detta.
Det enda som kan göras är att ta ansvar som svarar mot graviditetsplanering, att registreras och granskas i tid. Men igen, en sådan åtgärd skulle hellre inte vara profylaktisk, men kognitiv, som vilken undersökning som helst. Men unga föräldrar i förväg vet vad de ska förbereda sig för, och i fall av ett positivt svar kommer de att bestämma om de kommer att kunna ta på sig sådant ansvar som att höja ett sjukt barn.
Prognos
Prognosen för Anghelmans syndrom beror på karaktären av kromosomal abnormitet och tidpunkten för detektering. Den svåraste delen är för de barn vars 15 kromosomer innehåller "saknade" gener (radering). Sannolikheten att gå och prata i sådana patienter är extremt liten. De återstående fallen med ett uppmärksamt tillvägagångssätt och kärlek till ditt barn är mottagliga för korrigering.
Sådana patienter, tyvärr, kan inte bli fulla medlemmar i samhället, trots att de är långt ifrån dumma, de förstår talet och dess mening. Här är bara problem med kommunikation de har för livet. Patienter kan undervisas språk från barndomen, men man kan inte tvingas att kommunicera med ord. Leksikonet för "talande" patienter är begränsat till ett minimum av ord som används i vardagen (5-15 ord).
När det gäller livslängden och allmän hälsa hos patienter med Anghelmans syndrom fluktuerar siffrorna här i genomsnitt. Vid vuxen ålder upplever patienter i allmänhet hälsoproblem som skolios och fetma, som med rätt tillvägagångssätt för behandling inte är livshotande.