Medicinsk expert av artikeln

Nya publikationer
Xeroderma pigmentosum
Senast recenserade: 04.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
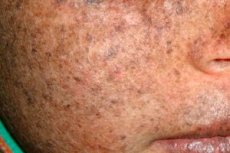
Xeroderma pigmentosum är en autosomal recessiv genetisk sjukdom som påverkar DNA-reparation. Sjukdomen orsakas av mutationer i gener som är involverade i reparationen av DNA som skadats av ultraviolett strålning och andra giftiga ämnen.
Oftast drabbar denna sjukdom barn, som också kallas "nattens barn". Vanliga komplikationer av sjukdomen är basalcellscancer och andra maligna neoplasmer i huden, metastaserande malignt melanom och skivepitelcancer.
 [ 1 ]
[ 1 ]
Patogenes
Brist på UV-endonukleasenzymer i patientens celler (i fibroblaster) eller deras fullständiga frånvaro spelar en viktig roll i sjukdomens utveckling. Detta enzym ansvarar för reproduktionen av DNA som skadats av ultravioletta strålar. Enligt andra källor spelar brist på DNA-polymeras-1-enzymet en viktig roll i sjukdomens utveckling hos vissa patienter. Sjukdomen utvecklas oftast under påverkan av strålar med en våglängd på 280-310 nm. Man tror att pigmentxerodermi utvecklas på grund av att olika fotodynamiska ämnen kommer in i kroppen eller en ökning av porfyriner i den mänskliga biologiska miljön.
I sjukdomens tidiga stadier observeras hyperkeratos, atrofi av epidermisfokus, uttunning av Malpighi-skiktet, en ökning av melaningranuler i cellbasalskiktet och kronisk inflammation som infiltrerar det övre lagret av dermis (främst runt blodkärl). Därefter upptäcks degenerativa förändringar i kollagen och elastiska fibrer, och i tumörstadiet avslöjas tecken som är karakteristiska för hudcancer.
Symtom xeroderma pigmentosum
Män och kvinnor drabbas lika mycket av denna sjukdom. Sjukdomen börjar i tidig barndom på våren eller sommaren. Patientens hud är särskilt känslig för solljus. Därför uppträder det första utslaget i form av hyperpigmentering, liknande en födelsemärke, på hudområden som utsätts för solljus. Utslagen ökar, erytem intensifieras, blir mörkbruna, telangiektasi, angiom och atrofi uppträder. Därefter växer papillom och vårtor (främst på huden i ansiktet och på halsen), fläckar och sår uppstår. Som ett resultat av sårärrbildning blir näsan tunnare ("fågelnäbb") och ögonlocket vrider sig. Papillom utvecklas vanligtvis till maligna tumörer. Pigmenterad xerodermi förekommer som regel tillsammans med spinocellulär cancer, melanosarkom.
Vad stör dig?
Stages
I det kliniska förloppet av xeroderma pigmentosum skiljer man sig åt i fem stadier: inflammatorisk (erytematös), hyperpigmentering, atrofi, hyperkeratos och maligna tumörer.
Under det inflammatoriska (erytematösa) stadiet uppstår svullnad, röda fläckar och ibland blåsor och vesiklar på hudområden som utsätts för solljus (ansikte, hals, övre delen av bröstet, armar, händer). Det finns nästan inga utslag på områden som inte utsätts för solljus.
Vid hyperpigmentering uppträder ljusbruna, bruna hyperpigmenterade fläckar som liknar födelsemärken i stället för de röda fläckarna.
Vid atrofi torkar huden ut, blir tunnare och rynkor bildas. Många små, stellatformade telangiektasier och ärr med en blank yta observeras på huden på läppar och näsa. På grund av atrofi och ärr utvecklas mikrotomi (minskning av munöppningen), ektropion, förtunning av öron och näsa, atresi i näsöppningen och munnen. Hos 80-85% av patienterna påverkas ögonen: konjunktivit, keratit, skador på hornhinnan och slemhinnan samt försvagning av synen observeras. Dyskromi, telangiektasi, hyperkeratotiska förändringar och tumörer observeras på huden på ögonlocken.
Vid hyperkeratos uppträder vårtiga tumörer, papillom, keratoakantom, fibrom, angiofibriom och andra godartade tumörer i de ovan beskrivna patologiska fokusen. Därför inkluderar vissa forskare pigmentxerodermi i gruppen av precancerösa sjukdomar.
Efter 10–15 år från sjukdomsdebut uppträder maligna hudtumörer (basaliom, endoteliom, angiosarkom) i atrofiskt förändrade fokus och pigmentfläckar. Tumörerna förstörs på kort tid, metastaserar till inre organ och leder till döden. I de inre organen och vävnaderna hos vissa patienter observeras generella dystrofiska förändringar (syndaktelier i andra och tredje tån, tanddystrofi, fullständigt håravfall etc.).
Formulär
Den neurologiska formen av xeroderma pigmentosum manifesterar sig i två syndrom.
Reeds syndrom kännetecknas av kliniska manifestationer av xeroderma pigmentosum och mikrocefali, idiopati och långsam tillväxt av skelettet. Vid sjukdomens neurologiska form är DNA-reparation svår och röntgenbehandling leder till en ökning av de huvudsakliga patologiska fokusen.
De Sinctis-X Cocchione syndrom kännetecknas av följande kliniska tecken:
- utveckling av kliniska tecken på xeroderma pigmentosum på särskilt ljuskänslig hud;
- tidigt uppträdande av maligna tumörer;
- samtidig förlopp av spastisk förlamning, mikrocefali och demens;
- medfödda missbildningar och dvärgväxt;
- underutveckling av gonaderna;
- frekventa missfall;
- recessiv överföring genom arv.
Enligt vissa dermatologer är Santis-Cacchione syndrom inte en oberoende sjukdom, utan en allvarlig och fullt uttryckt klinisk manifestation av xeroderma pigmentosum.
Genetiska former
Typer |
Gen |
Ställe |
Beskrivning |
Typ A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) grupp A – klassisk form |
Typ B, II, XPB |
XPB |
2q21 |
XP Grupp B |
Typ C, III, XPC |
XPC |
3p25 |
XP Grupp C |
Typ D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP Grupp D eller De Sanctis-Cacchione syndrom |
Typ E, V, XPE |
DDB2 |
23:00-12:00 |
XP Grupp E |
Typ F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
XP Grupp F |
Typ G, VII, XPG |
RAD2ERCC5 |
13q33 |
XP grupp G och COFS syndrom (cerebro-okulo-facioskeletalt syndrom) typ 3 |
Typ V, XPV |
POLH |
6p21.1-p12 |
Variant Xeroderma Pigmentosa |
Vad behöver man undersöka?
Hur man undersöker?
Vem ska du kontakta?
Behandling xeroderma pigmentosum
Antimalariamedel (delagyl, plaquenil, resoquin, etc.), som skyddar DNA från ultravioletta strålar, hämmar depolymerisationsprocessen, minskar hudens känslighet (fotosensibilisering) för solljus. Dessa läkemedel har antiinflammatoriska och hyposensibiliserande egenskaper. Det rekommenderas att genomföra allmän terapi tillsammans med vitaminbehandling (B1, B2, PP, B6, B12, A, E), antihistaminer (tavegil, difenhydramin, suprastin) och desensibiliserande medel (natriumtiosulfat, 10 % kalciumklorid intravenöst 10 ml).
För lokal behandling används solskyddskrämer och salvor.
Vid tumörformen av pigmentxerodermi används kirurgiska metoder, flytande kväve och laserstrålar. För att skydda dig mot solljus måste du bära löst sittande kläder, solhatt och handskar.
Prognos
De flesta patienter (2/3) dör innan de når 15 års ålder. Vissa patienter kan leva i 40–50 år.
[ 20 ]