Medicinsk expert av artikeln

Nya publikationer
Angelmans syndrom hos barn och vuxna
Senast recenserade: 04.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.

Det finns ett antal sjukdomar för vilka uttryck som "ta hand om dig så blir du inte sjuk" låter minst sagt löjliga. Det här är patologier där vissa psykiska och fysiska avvikelser är inneboende i barnets kropp redan före födseln, men föräldrarna är inte att skylla för detta. Sådana sjukdomar orsakas av mutationer eller avvikelser i kromosomuppsättningar och kallas kromosomala eller genetiska. Angelmans syndrom, Downs syndrom, Pataus syndrom, Edwards syndrom, Turners syndrom, Prader-Willis syndrom - detta är bara en del av de genetiska sjukdomarna från en ganska hyfsad lista.
Glad man-syndrom
Den här gången ska vi prata om patologin uppkallad efter den engelske barnläkaren Harry Angelman, som först tog upp frågan om detta problem 1965, efter att dagen innan ha stött på tre ovanliga barn i sin praktik, förenade av gemensamma säregna symtom. Läkaren kallade dessa barn för dockbarn och skrev en artikel om dem, som ursprungligen hette "Barn-marionetter". Själva artikeln och dess titel skrevs under intryck av en målning som setts på ett av museerna i Verona. Målningen avbildade en skrattande pojke, och den kallades "Dockpojken". Associationen mellan barnet som avbildas i målningen och de tre barn som Angelman en gång stötte på i sin praktik fick barnläkaren att slå samman barnen i en grupp på grund av den sjukdom de hade.
Det är inget förvånande i att barnen som nämns i artikeln inte uppmärksammades av andra läkare. Vid första anblicken verkade det ju som att de hade helt olika sjukdomar, så olika var den allmänna kliniska bilden av sjukdomen i tre olika fall. Kanske skulle den "nya" kromosompatologin ha intresserat andra forskare, men vid den tiden var genetiken ännu inte tillräckligt utvecklad för att bekräfta den engelske läkarens hypotes. Därför, efter ett visst intresse för den, kastades artikeln på bakre hyllan under lång tid.
Nästa omnämnande av Angelmans syndrom, vilket är vad artikeln av den engelske barnläkaren G. Angelman nu kallades, går tillbaka till början av 80-talet av 1900-talet. Och först 1987 kunde man hitta orsaken till att en liten del av barnen föds med sådana avvikelser att de utifrån sett verkar ständigt le och vara glada. Faktum är att detta inte alls är sant, och leendet är bara en grimas, bakom vilken döljer sig en olycklig mänsklig själ och föräldrarnas smärta.
Epidemiologi
Enligt statistik kan en kromosommutation hos ett barn utvecklas både mot bakgrund av liknande mutationer hos föräldrar och i frånvaro av sådana. Det finns ingen tydlig ärftlig karaktär av Angelmans syndrom (AS), men sannolikheten för att utveckla patologi hos föräldrar med kromosommutationer är ganska hög.
Det är också intressant att om en familj redan har ett barn med AS, finns det en procents chans att få ett andra barn med samma sjukdom, även om föräldrarna är friska.
Det finns fortfarande ingen exakt statistik över antalet patienter med Angelmans syndrom. Orsaken är kanske variationen i symtom, som kan förekomma i en viss sammansättning eller helt utebli under en längre tid. Det antas att sjukdomens prevalens är: 1 barn per 20 000 nyfödda. Men denna siffra är mycket ungefärlig.
Orsaker Angelmans syndrom
Angelmans syndrom är ett medicinskt namn för en kromosomal patologi, men det är långt ifrån det enda. Folk kallar denna sjukdom dockbarnssyndrom, glad dockasyndrom, Petrusjkas syndrom och skrattande dockasyndrom. Folk kommer på alla möjliga namn (ibland till och med stötande för patienterna själva och deras föräldrar), men en sjukdom är en sjukdom, oavsett hur rolig den kan se ut och oavsett orsakerna.
Och orsakerna till utvecklingen av Angelmans syndrom, liksom många andra genetiska patologier, är i alla fall störningar i strukturen hos en av kromosomerna eller kromosomuppsättningen som helhet. Men i vårt fall ligger hela problemet i kromosom 15, som ärvs från modern. Det vill säga, faderns kromosom har i detta fall inga avvikelser, men den kvinnliga genomgår vissa mutationer.
Beroende på typen av kromosomavvikelse klassificeras Angelmans syndrom som en kromosomal mutation. Sådana mutationer anses vara:
- En deletion (avsaknad av en del av en kromosom som innehåller en viss uppsättning gener; om en av generna saknas talar vi om en mikrodeletion), vilket är resultatet av två brott och en återförening, när en del av den ursprungliga kromosomen går förlorad.
- Duplicering (närvaron av en extra sektion i en kromosom som är en kopia av en befintlig), vilket i de flesta fall leder till en persons död och mer sällan till infertilitet.
- Inversion (omvändning av en av kromosomens sektioner med 180 grader, dvs. i motsatt riktning, och då är generna i den placerade i motsatt ordning), när kromosomens trasiga ändar är sammankopplade i en annan ordning än originalet.
- Insättning (om en del av det genetiska materialet i en kromosom är felplacerat),
- translokation (om en viss del av en kromosom är fäst vid en annan kromosom; en sådan mutation kan vara ömsesidig utan förlust av sektioner).
Om barnet får en muterad kromosom från en intet ont anande mor är det dömt att födas med missbildningar. Den vanligaste orsaken till Angelmans syndrom anses fortfarande vara en deletion av moderns 15:e kromosom, när en liten del saknas. Mindre vanliga mutationer i "skrattdockesyndromet" anses vara:
- translokation,
- unipaternal disomi (om barnet fick ett par kromosomer från fadern, saknas moderns kromosom),
- mutation av gener i DNA, vilka både är det huvudsakliga byggmaterialet (genetiska) och instruktioner för dess korrekta användning (i synnerhet mutation av ube3a-genen i moderns kromosom).
Förekomsten av en av dessa mutationer hos föräldrarna är en riskfaktor för utveckling av Angelmans syndrom hos barn. Men inte bara kromosomala mutationer, utan även genomiska (som är förknippade med en kvantitativ förändring i kromosomuppsättningar och är vanligare än kromosomala) kan provocera utvecklingen av sjukdomen hos ett barn. Vanliga genomiska mutationer inkluderar kromosomtrisomi (om en persons kromosomuppsättning har mer än 46 kromosomer).
För att en patologi ska uppstå hos ett barn är det inte alls nödvändigt att föräldrarna har kromosomavvikelser. Och ändå finns det en viss andel patienter vars sjukdom är ärftlig.
Patogenes
Låt oss fördjupa oss lite i biologi, eller mer exakt, genetik. Den genetiska informationen för varje enskild mänsklig organism finns i 23 kromosompar. En kromosom från ett par överförs till barnet från fadern, den andra från modern. Alla kromosompar skiljer sig åt i form och storlek och bär på viss information. Således är det 23:e kromosomparet (X- och Y-kromosomer) ansvarigt för bildandet av barnets sexuella egenskaper (XX - flicka, XY - pojke, medan Y-kromosomen bara kan tas emot av barnet från fadern).
Idealiskt sett får ett barn 46 kromosomer från sina föräldrar, vilka bildar barnets genetiska egenskaper och förutbestämmer det som individ. Ett större antal kromosomer kallas trisomi och anses vara en avvikelse från normen. Till exempel orsakar närvaron av kromosom 47 i kromosomuppsättningen (karyotyp, artbestämmande och individuella egenskaper) uppkomsten av Downs syndrom.
Om kromosomerna färgas med ett speciellt färgämne kan man under mikroskopet se ränder i olika nyanser längs var och en av dem. Inuti varje rand finns ett stort antal gener. Alla dessa ränder är numrerade av forskare och har en fast plats. Avsaknaden av en av randarna anses vara en avvikelse från normen. Vid Angelmans syndrom kan man mycket ofta observera avsaknaden av segment av moderns kromosom i intervallet q11-q13, belägna i den långa armen, vars antal DNA-baser endast är cirka 4 miljoner.
Huvudkomponenten i kromosomen anses vara en otroligt lång DNA-molekyl som innehåller tusentals gener och tiotals och hundratals miljoner kvävebaser. Således innehåller kromosom 15, som är ansvarig för utvecklingen av Angelmans syndrom och flera andra, 1200 gener och cirka 100 miljoner baser. Eventuella störningar i DNA-molekylens struktur kommer säkerligen att påverka det blivande barnets utseende och utveckling.
Den genetiska informationen i gener omvandlas till protein eller RNA. Denna process kallas genuttryck. På så sätt får den genetiska informationen som tas emot från föräldrarna både form och innehåll, vilket förkroppsligas i deras unika kvinnliga eller manliga arvinge.
Det finns ett antal patologier med en icke-klassisk typ av arv, inklusive Angelmans syndrom, där gener som erhållits från föräldrar som en del av parade kromosomer bär ett unikt avtryck av föräldrarna och manifesterar sig på olika sätt.
Så, Angelmans syndrom är ett slående exempel på genomisk prägling, där genuttryck i barnets kropp är direkt beroende av vilken förälder allelerna erhölls från (olika former av samma gen, mottagna från fadern och modern, belägna på identiska sektioner av parade kromosomer). Det vill säga, endast avvikelser i moderns kromosom leder till utvecklingen av syndromet, medan mutationer och strukturella störningar i faderns kromosom orsakar helt olika patologier.
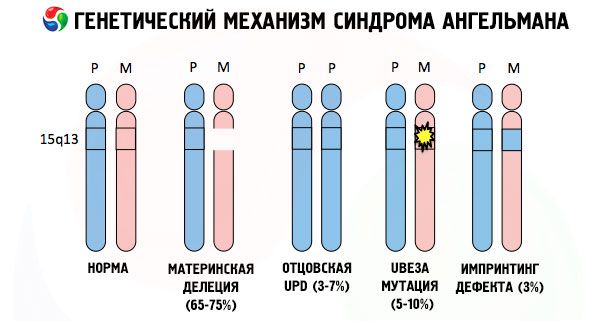
Vid denna patologi saknas vissa gener i moderns kromosom eller så finns det en förlust/minskning av aktiviteten hos enskilda gener (i de allra flesta fall ube3a-genen, som är involverad i metabolismen av ubiquitin, ett protein som reglerar nedbrytningen av andra proteiner). Som ett resultat diagnostiseras barnet med psykiska utvecklingsavvikelser och fysiska missbildningar.
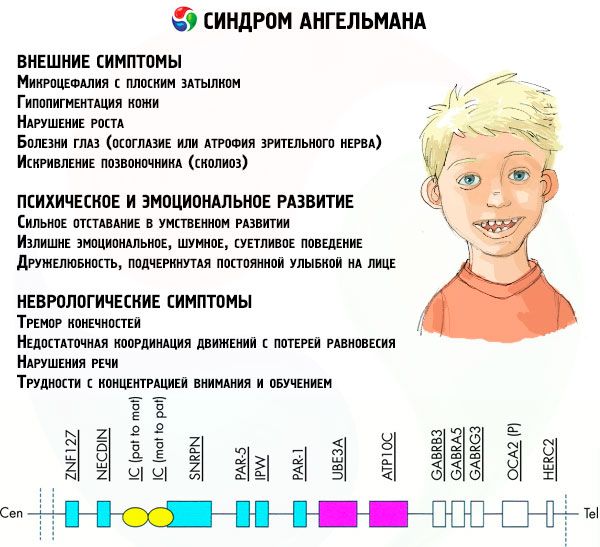
Symtom Angelmans syndrom
Symtomen på Angelmans syndrom påverkar olika aspekter av ett barns liv och utveckling: fysiska, neurologiska, mentala. Baserat på detta kan 3 grupper av symtom identifieras som indikerar utvecklingen av denna patologi.
- Externa eller fysiska symtom:
- ett oproportionerligt litet huvud jämfört med kroppen och lemmarna, som är av normal storlek,
- för bred mun,
- det finns nästan alltid ett leende på läpparna (med öppen mun),
- glesa tänder,
- smal överläpp,
- ofta utskjutande bred tunga,
- utskjutande underkäke,
- spetsig haka,
- mycket ljus hud, ofta hår (albinism, förknippad med att kroppen inte producerar pigmentet melanin),
- mörka fläckar på ljus hud (hypopigmentering på grund av otillräcklig melaninproduktion)
- fysiska eller yttre symtom: ögonsjukdomar såsom strabismus eller synnervsatrofi,
- krökning av ryggraden (skolios),
- stela ben (när man går böjer en person inte benen vid knäna på grund av låg rörlighet i lederna, därav jämförelsen med en dockas gång).
- Symtom relaterade till mental och emotionell utveckling:
- allvarlig psykisk utvecklingsstörning,
- alltför känslosamt, högljutt, kinkigt beteende,
- frekvent klappning av händer,
- uttryckt vänlighet, betonad av ett ständigt leende på läpparna,
- frekvent skratt utan anledning.
- Neurologiska symtom:
- tremor i lemmarna,
- otillräcklig koordination av rörelser med förlust av balans,
- minskad muskeltonus,
- olika sömnstörningar,
- frekventa hysteriska anfall i barndomen,
- talstörningar (barnet börjar prata sent, har dålig kommunikationsförmåga och sluddrigt tal),
- hyperaktivitet mot bakgrund av ökad excitabilitet,
- svårigheter att koncentrera sig och lära sig.
Men detta är en generaliserad bild av sjukdomen. Faktum är att den kliniska bilden av Angelmans syndrom till stor del beror på sjukdomens utvecklingsstadium och vilken typ av kromosommutation som orsakade patologin. Detta innebär att sjukdomens symtom kan skilja sig avsevärt hos olika patienter, vilket under lång tid inte tillät oss att skilja patologin från andra med en liknande klinisk bild.
Bland det totala antalet symtom kan vi lyfta fram de som är karakteristiska för alla patienter utan undantag:
- allvarlig psykisk utvecklingsstörning,
- olämpligt beteende (orimligt skratt, ökad upphetsning, dålig koncentration, eufori),
- underutveckling av motoriska färdigheter,
- dålig koordination av rörelser, gångataxi (ojämn takt, svajande från sida till sida etc.), tremor i extremiteterna.
- talutvecklingsstörning med övervikt av icke-verbala kommunikationsmedel.
Bland de symtom som den stora majoriteten av patienterna upplever kan följande urskiljas:
- oproportioner mellan huvud och kropp orsakade av försenad fysisk utveckling,
- hos många patienter är skallens form sådan att hjärnans storlek förblir mindre än hos friska personer (mikrocefali),
- epileptiska anfall före 3 års ålder med en progressiv minskning av styrka och frekvens vid högre ålder,
- förvrängning av EEG-parametrar (fluktuationer och hög amplitud hos lågfrekventa vågor).
Dessa symtom är ganska vanliga, men 20 % av patienterna med Angelmans syndrom har dem inte.
Ännu mindre ofta är det möjligt att diagnostisera sådana manifestationer av sjukdomen som:
- svår eller mild strabismus,
- dålig kontroll över tungans rörelser, vilket resulterar i att patienter ofta sticker ut tungan utan anledning,
- svårigheter att svälja och suga, särskilt hos små barn,
- störningar i hud- och ögonpigmentering,
- armarna upplyfta eller böjda under gång,
- hyperreflexi,
- sömnstörningar, särskilt i barndomen,
- frekvent salivutsöndring,
- omättlig törst,
- överaktiva tuggrörelser,
- överkänslighet mot värme,
- platt bakhuvud,
- utskjutande underkäke,
- släta handflator.
En ganska stor andel patienter har problem med urinering, som de dåligt kontrollerar, nedsatt finmotorik, vilket skapar svårigheter med egenvård och inlärning, samt övervikt. Nästan alla patienter upplever puberteten senare än friska jämnåriga.
Barn med Angelmans syndrom uppfattar muntligt tal väl och förstår det, men vill inte delta i samtal och begränsar sitt tal till flera dussin ord som är nödvändiga i vardagen. Men i vuxen ålder ser sådana patienter yngre ut än sina jämnåriga utan genetiska patologier.
Många symtom på Angelmans syndrom är inkonstanta, så den kliniska bilden av sjukdomen förändras avsevärt med åldern. Konvulsioner och epileptiska anfall blir mindre frekventa eller försvinner helt och hållet, patienten blir mindre upphetsad och sömnen förbättras.
Komplikationer och konsekvenser
Angelmans syndrom är en allvarlig, för närvarande praktiskt taget obotlig kromosomal patologi som berövar patienter möjligheten att leva ett normalt liv. Hur livet för ett barn med AS kommer att vara beror till stor del på typen av kromosomavvikelse.
Duplicering av ett kromosomsegment är i de flesta fall oförenligt med liv. Och även om sådana patienter inte dör i spädbarnsåldern och når puberteten, har de ingen chans att få barn.
Bortfallet eller avsaknaden av en del av de gener som oftast förekommer vid Angelmans syndrom är ett hinder för barnet att lära sig gå och prata. Sådana barn har en allvarligare form av utvecklingsstörning, och epileptiska anfall förekommer oftare, och deras intensitet är mycket större än hos patienter med andra kromosomavvikelser.
Om det bara finns en mutation av en gen, kan barnet med tillräcklig uppmärksamhet och tillvägagångssätt lära sig grunderna i egenvård, kommunikation och interaktion i grupp, även om det fortfarande kommer att halka efter sina jämnåriga i utveckling.
För barn med Angelmans syndrom, som är vänliga av naturen, är det viktigaste föräldrarnas kärlek och uppmärksamhet. Endast i detta fall kommer barnets utbildning att bära frukt, även om den är liten. Naturligtvis kommer patienter med AS inte att kunna studera i en vanlig skola. De behöver specialklasser där barn först lär sig att koncentrera sig, och sedan gradvis får de grunderna i skolkunskap.
Diagnostik Angelmans syndrom
Angelmans syndrom är en medfödd utvecklingspatologi. Men på grund av vissa omständigheter är det ofta omöjligt att diagnostisera det i spädbarnsåldern och tidig barndom. Detta beror på ospecificiteten och svaga symtom hos spädbarn och barn under 3 år. Och sjukdomens förekomst i vårt land är inte så stor att läkare har lärt sig att känna igen den bland sina kollegor.
Angelmans syndrom hos spädbarn kan manifestera sig som minskad muskeltonus, vilket yttrar sig i problem med matning (svaghet i sug- och sväljningsreflexen) och senare svårigheter att lära sig gå (sådana barn börjar gå mycket senare). Dessa symtom är de första tecknen på en utvecklingsavvikelse hos barnet, vilket mycket väl kan vara förknippat med en kromosomavvikelse. Endast genetisk analys kan bekräfta detta antagande.
Särskild uppmärksamhet ägnas åt barn vars föräldrar har olika genomiska eller kromosomala sjukdomar. Sjukdomen kanske trots allt inte manifesterar sig till en början, och om patologin upptäcks i tid, genom att börja arbeta intensivt med barnet, är det möjligt att uppnå betydligt större framgång i inlärningen, vilket saktar ner sjukdomsprogressionen.
Om föräldrarna har olika kromosomavvikelser utförs genetisk analys redan innan barnet föds, eftersom SA är en av de patologier som kan upptäckas i embryonalstadiet.
Insamling av material för genetisk forskning kan utföras på två sätt:
- invasiv (med en viss procentuell risk, eftersom det är nödvändigt att penetrera livmodern för att ta ett prov av fostervätska),
- icke-invasiv (analys av barnets DNA från moderns blod).
Följande studier genomförs sedan:
- fluorescerande in situ-hybridisering (FISH-metoden) – bindning av en DNA-sond märkt med ett speciellt färgämne till det DNA som studeras, följt av undersökning i mikroskop.
- analys av mutationer i ube3a-genen och imprintade gener,
- DNA-metyleringsanalys med hjälp av speciella metoder som används inom genetik.
Genetiska tester ger ganska noggrann information vid kromosomavvikelser, vilket innebär att blivande föräldrar vet i förväg vad de ska förbereda sig för. Det finns dock undantag. Hos en viss patientgrupp, i närvaro av alla symtom som indikerar patologi, förblir testresultaten normala. Det vill säga, patologi kan endast identifieras genom att noggrant observera barnet från tidig barndom: hur det äter, när det började gå och prata, om det böjer benen när det går, etc.
Förutom FISH-metoden kan man bland de instrumentella diagnostiska metoderna för Angelmans syndrom skilja ut tomografi (CT eller MRI), som hjälper till att bestämma hjärnans tillstånd och storlek, och ett elektroencefalogram (EEG), som visar hur enskilda delar av hjärnan fungerar.
Läkare ställer vanligtvis en slutgiltig diagnos vid 3-7 års ålder, när patienten redan har de flesta symtomen och dynamiken i sjukdomsutvecklingen är synlig.
Vilka tester behövs?
Differentiell diagnos
Angelmans syndrom är en genetisk patologi som i princip inte har några specifika manifestationer. De flesta symtom kan lika gärna tyda på både AS och andra genetiska patologier.
Differentialdiagnos av Angelmans syndrom utförs med följande patologier:
- Pitt-Hopkins syndrom (patienter kännetecknas av utvecklingsstörning, gladlynt karaktär, leende, de har en ganska stor och bred mun, mikrocefali noteras). Skillnaden är attacker av hyperventilation och andningshållning i vaket tillstånd.
- Christiansons syndrom (patienter är psykiskt funktionsnedsatta personer med ett glatt sinnelag, oförmögna att tala, kännetecknade av mikrocefali, ataxi, kramper, ofrivilliga muskelrörelser).
- Mowat-Wilsons syndrom (symtom: utvecklingsstörning, epileptiska anfall, spetsig haka, öppen mun, glatt ansiktsuttryck, mikrocefali). Särskiljningssätt: stort avstånd mellan ögonen, ögonen lutade inåt, rundad nässpets, bakåtvänd öronmussla.
- Kabuki-syndrom (kännetecknas av mild till måttlig utvecklingsstörning, tal- och motoriska problem, muskelsvaghet, epileptiska anfall, mikrocefali, långa intervall mellan klåda och nedsatt koordination). Kännetecknas av välvda ögonbryn, utåtböjd laterala del av det nedre ögonlocket, brett sittande ögon, långa palpebrala fissurer med långa, tjocka ögonfransar.
- Retts syndrom (differentiering från AS hos kvinnor). Symtom: försenad talutveckling, kramper, mikrocefali. Skillnaden är att det inte finns något glatt uttryck i ansiktet, det förekommer attacker av apné och apraxi, vilket fortskrider med tiden.
- Autosomalt recessivt mentalt tardationssyndrom 38 (symtom: markant utvecklingsstörning med förseningar i motoriska färdigheter och tal, muskelsvaghet, ätproblem i spädbarnsåldern, impulsivitet). Kännetecken är irisens blå färg.
- MECP 2-genduplikeringssyndrom (differentiering från SA hos män). Symtom: svår utvecklingsstörning, muskelsvaghet sedan barndomen, talproblem eller talbrist, epilepsi. Särskiljningar: progressiv myopati, ständigt återkommande infektioner.
- Kleefstras syndrom (symtom: tal- och tankesvårigheter, muskelsvaghet, sömnstörningar, bristande uppmärksamhet, öppen mun, hyperaktivitet, kramper, ataxi, balansstörningar). Särskilda drag: platt ansikte, kort trubbig näsa, brett sittande ögon, stor utåtböjd underläpp, aggressiva utbrott.
- Smith-Magenis syndrom (kännetecknas av kramper, sömnproblem, intellektuella och motoriska utvecklingsstörningar). Utmärkande drag inkluderar ett brett och platt ansikte och en framträdande panna.
- Koolen-de Vries syndrom (mild till måttlig utvecklingsstörning, muskelsvaghet, kramper, vänlighet). Kännetecken: långt ansikte med hög panna, utstående öron, sneda ögon, hög ledrörlighet, medfödda hjärtfel.
- Phelan-McDermids syndrom (symtom: utvecklingsstörning, talsvårigheter eller brist på talförmåga). Särskilda kännetecken: stora händer med utvecklade muskler, muskelsvaghet från födseln, svag svettning.
Sådana patologier som adenylsuccinatbrist, autosomalt recessivt mental retardationssyndrom 1, kromosom 2q23.1-dupliceringssyndrom, FOXG1-, STXBP1- eller MEF2C-genhaploinsufficienssyndrom och några andra kan "skryta" med symtom som liknar Angelmans syndrom.
Läkarens uppgift är att ställa en korrekt diagnos, skilja Angelmans syndrom från patologier med liknande symtom och förskriva effektiv behandling som är relevant för det diagnostiserade skedet av sjukdomen.
Vem ska du kontakta?
Behandling Angelmans syndrom
Angelmans syndrom är en av de patologier för vilka medicinen fortfarande söker effektiv behandling. Den etiologiska behandlingen av sjukdomen är i utvecklingsstadiet med olika metoder och medel, av vilka många ännu inte har testats på människor. Detta innebär att läkare för närvarande måste begränsa sig till symptomatisk behandling, vilket på något sätt hjälper till att lindra den föga avundsvärda situationen för barn och vuxna med marionettsyndrom, som lider av epileptiska anfall, salivutsöndring, hypotoni och sömnstörningar.
Det är således möjligt att minska frekvensen och styrkan av epileptiska anfall med hjälp av ett korrekt valt antikonvulsivt läkemedel. Men hela svårigheten är att anfall hos patienter med SA skiljer sig från vanliga epileptiska anfall genom att de kännetecknas av flera typer av anfall, vilket innebär att tillståndet kan lindras genom att administrera flera läkemedel samtidigt.
De mest populära antikonvulsiva läkemedlen som används för att behandla Angelmans syndrom är: valproinsyra, topiramat, lamotrigin, levetiracetam, klonazepam och läkemedel baserade på dem. Mindre vanligt förekommande är läkemedel baserade på karmazepin, fenytoin, fenobarbital och etosuximid, eftersom vissa av dem kan framkalla en paradoxal effekt som består i att förstärka och öka frekvensen av epileptiska anfall. Detta händer om läkemedlet används som en del av monoterapi.
För att behandla dregling används vanligtvis två metoder: medicinska (läkemedel som hämmar salivproduktionen) och kirurgiska metoder, som innebär reimplantation av spottgångarna. Men vid SA anses dessa metoder vara ineffektiva, och frågan förblir öppen. Föräldrar och de som vårdar sådana patienter måste ägna särskild uppmärksamhet åt denna fråga, eftersom patienterna själva vanligtvis inte kontrollerar dreglingen, och vissa helt enkelt inte kan ta hand om sig själva.
Ett annat problem är kort sömntid. Ofta sover barn med Angelmans syndrom högst 5 timmar, vilket har en negativ inverkan på hela kroppens funktion. Lättretliga, aktiva barn som älskar lekar och kommunikation (även om de försöker begränsa sig till icke-verbala metoder) är märkbart trötta under dagen. För att få en god vila behöver kroppen en djup, fullständig sömn, men det är just detta som är haken.
Det verkar som att lugnande läkemedel (fentiaziner och atypiska antipsykotika) som lugnar nervsystemet borde vara tillräckliga för att förbättra sömnen hos retbara patienter. Men vid AS är användningen av sådana läkemedel förenad med förekomsten av negativa effekter. Därför föredrar läkare fortfarande milda sömntabletter, såsom melatonin (ett naturligt hormonellt läkemedel baserat på sömnhormonet), som ges till patienter en timme före sänggåendet i mängden 1 tablett, och difenhydramin. Administreringsfrekvensen och doseringen bestäms av läkaren beroende på patientens tillstånd och ålder.
Ibland har patienter med Angelmans syndrom problem med matsmältning och avföring. Du kan förbättra din avföring med laxermedel (helst naturläkemedel).
Eller så kan man närma sig problemet annorlunda, som amerikanska läkare gjorde, baserat på vissa metoder för att behandla autism, eftersom många symtom som är karakteristiska för AS också är karakteristiska för autism (impulsivitet, ofrivilliga rörelser, repetitiva handlingar, uppmärksamhetsbrist, kommunikationsproblem, etc.). Det noterades att införandet av hormonet sekretin, som normaliserar matsmältning och avföring, har en positiv effekt på patienternas uppmärksamhet, och oxytocin hjälper till att förbättra barnets kognitiva förmågor och minne, och korrekt beteende.
Visst räcker inte hormoner ensamma, särskilt inte när det gäller barn. Vid Angelmans syndrom är beteendeterapi, arbete med en psykolog och logoped (undervisning i icke-verbala kommunikationsmetoder och teckenspråk) indicerat. Utbildningen av sådana barn bör baseras på ett individuellt program med deltagande av specialutbildade lärare, en psykolog och föräldrar. Tyvärr är detta inte möjligt överallt, och familjer lämnas ensamma med sina problem.
Eftersom många unga patienter med AS lider av låg muskeltonus och ledproblem ägnas stor uppmärksamhet åt fysioterapi. Oftast tillgriper läkare användningen av paraffinbehandling, elektrofores och magnetisk terapi.
Aktiv tonisk massage och speciella övningar inom terapeutisk fysisk träning hjälper det sjuka barnet att stå på fötter och gå med självförtroende efter ett tag. Vattengymnastik är särskilt användbart i detta avseende, vilket rekommenderas för SA i kallt vatten. Det ökar muskeltonus och lär barnet att kontrollera sin kropp och koordinera rörelser.
Antikonvulsiv behandling
Det farligaste symptomet på Angelmans syndrom är anfall som liknar de vid epilepsi. Detta symptom observeras hos 80 % av patienterna, vilket innebär att alla behöver ordineras effektiv antikonvulsiv behandling.
Behandling av epileptiska anfall utförs med hjälp av vitaminer och antikonvulsiva medel. Vid Angelmans syndrom, åtföljt av konvulsivt syndrom, kommer vitaminer i grupp B, samt vitamin C, D och E, att vara användbara. Men att förskriva vitaminbehandling på egen hand i detta fall är mycket farligt, eftersom okontrollerat intag av vitaminer kan minska effekten av antiepileptiska läkemedel och framkalla nya, allvarligare och längre anfall.
Valet av antikonvulsiva läkemedel och förskrivningen av deras effektiva dosering bör också göras av en specialistläkare. Han eller hon avgör också om ett läkemedel räcker eller om patienten måste ta två eller fler läkemedel under en längre tid.
För de flesta patienter ordinerar läkare valproinsyraläkemedel (Valproinsyra, Depakine, Convulex, Valparin, etc.), vilket förhindrar anfall och förbättrar patienternas humör och mentala tillstånd.
Valproinsyra finns i form av tabletter, sirap och injektionslösningar. Det mest populära läkemedlet är depotläkemedlet "Depakine" i tabletter och som lösning för intravenös administrering. Doseringen av läkemedlet bestäms av läkaren individuellt beroende på patientens vikt, ålder och tillstånd.
Läkemedlet tas i samband med måltider 2 till 3 gånger per dag. Den genomsnittliga dagliga dosen är 20–30 mg per kilogram patientens vikt, den maximala dosen är 50 mg/kg per dag.
Kontraindikationer för användning. Använd inte vid lever- och bukspottkörteldysfunktion, hemorragisk diates, hepatit, porfyri och överkänslighet mot läkemedlet.
Biverkningar inkluderar handskakningar, matsmältnings- och avföringsproblem och förändringar i kroppsvikt.
"Topiramat" är också ett läkemedel som föredras vid SA. Det tillverkas i tablettform och används både som en del av monoterapi och i kombination med andra läkemedel.
Administreringssätt och dosering. Ta tabletterna oralt oavsett födointag. Den initiala dagliga dosen för vuxna är 25–50 mg, för barn – 0,5–1 mg/kg. Dosen ökas varje vecka enligt läkarens anvisningar.
Läkemedlet ska inte tas under graviditet och amning, samt vid överkänslighet mot dess komponenter. Läkemedlet har många olika biverkningar.
Läkemedel som en läkare kan ordinera för Angelmans syndrom: Klomazepam, Rivotril, Lamotrigin, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra, etc.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Traditionell medicin och homeopati
Traditionell medicin, liksom homeopatiska preparat, är naturligtvis relativt säker, men effektiviteten av sådan behandling för Angelmans syndrom kan anses vara kontroversiell.
Även om folkmedicin fortfarande kan hjälpa till med vissa saker, talar vi om att stoppa epileptiska anfall. I detta avseende kan örtbehandling vara ganska effektiv.
En god effekt ges av en medicinsk samling baserad på pion, lakrits och andmat (komponenterna tas i lika stora mängder). Örterna måste malas till mjöl. Efter 2 veckor från början av intaget kan du märka en signifikant minskning av anfallsfrekvensen.
Lavendelavkok (1 tesked per glas kokande vatten) är också användbart vid kramper. Blandningen kokas i 5 minuter och låt dra i en halvtimme. Medicinen tas på natten i 14 dagar.
En vattenhaltig (eller alkoholhaltig) infusion av morört anses effektiv för epileptiska anfall.
Av de homeopatiska preparaten för att förebygga anfall vid Angelmans syndrom kan man använda läkemedel baserade på kamomill och morört, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Men det bör beaktas att endast en homeopatisk läkare kan ordinera effektiva och säkra doser av läkemedel i varje specifikt fall.
Förebyggande
Som läsaren förmodligen redan har förstått kan medicinen ännu inte förhindra genmutationer och andra kromosomavvikelser, samt korrigera situationen. Detta kan hända vem som helst, eftersom barn med Angelmans syndrom föds av friska föräldrar, och genetik, som för närvarande är en av de minst studerade grenarna inom medicin, kan ännu inte förklara detta.
Det enda man kan göra är att ta ett ansvarsfullt förhållningssätt till graviditetsplanering, registrera sig och genomgå undersökningar i tid. Men återigen, en sådan åtgärd kommer att vara mer pedagogisk än förebyggande, precis som alla undersökningar. Men unga föräldrar kommer att veta i förväg vad de ska förbereda sig för, och i händelse av ett positivt svar kommer de att avgöra om de kan ta på sig ett sådant ansvar som att uppfostra ett sjukt barn.
Prognos
Prognosen för Angelmans syndrom beror på kromosomavvikelsens natur och hur snabbt den upptäcks. De barn vars kromosom 15 innehåller "luckor" i gener (deletion) drabbas hårdast. Sannolikheten för att sådana patienter ska kunna gå och prata är extremt låg. Andra fall kan korrigeras med en noggrann strategi och kärlek till ditt barn.
Tyvärr kommer sådana patienter inte att kunna bli fullvärdiga samhällsmedlemmar, trots att de är långt ifrån dumma, de förstår tal och dess betydelse. De kommer dock att ha problem med kommunikationen resten av sina liv. Patienter kan lära sig teckenspråk från barndomen, men de kan inte tvingas att kommunicera med hjälp av ord. Ordförrådet hos "talande" patienter är begränsat till ett minimum av ord som används i vardagen (5-15 ord).
När det gäller förväntad livslängd och allmän hälsa för patienter med Angelmans syndrom fluktuerar siffrorna här kring genomsnittsvärden. I vuxen ålder drabbas patienter oftast av hälsoproblem som skolios och fetma, vilka med rätt behandlingsmetod inte är livshotande.