Medicinsk expert av artikeln

Nya publikationer
Alkaptonuri är en medfödd enzymavvikelse
Senast recenserade: 04.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
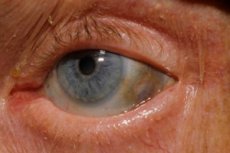
En av de mycket sällsynta metabola sjukdomarna, alkaptonuri, hänvisar till medfödda missbildningar i metabolismen av aminosyran tyrosin.
Detta syndrom kan också kallas homogentisatoxidasbrist, homogentisinuri, ärftlig ochronos eller svarturinsjukdom.[ 1 ]
Epidemiologi
Enligt statistiken finns det högst nio fall av alkaptonuri per 1 miljon människor. Och i de flesta europeiska länder finns det ett fall per 100–250 tusen levande födda.
Bland europeiska länder är undantaget Slovakien (särskilt den relativt lilla nordvästra regionen), där prevalensen av alkaptonuri är ett fall per 19 000 nyfödda. Detta beror troligen på att bland de slovakiska romska familjerna som bor där är graden av inavel (äktenskap mellan kusiner) den högsta i Europa: 10–14 %. [ 2 ]
Orsaker alkaptonuri
De exakta orsakerna till alkaptonuri, som en medfödd sjukdom i katabolismen (metabolisk nedbrytning) av den aromatiska (homocykliska) α-aminosyran tyrosin, har fastställts: denna typ av metabolisk sjukdom är en konsekvens av homozygota eller sammansatta heterozygota mutationer av en av de tusentals generna på kromosom 3, mer exakt, HGD-genen vid lokus 3q21-q23 på kromosomens långa arm. Denna gen kodar för nukleotidsekvenserna för leverenzymet homogentisat-1,2-dioxygenas [ 3 ] (även kallat homogentisinsyraoxidas eller homogentisatoxidas) - ett järnhaltigt metalloprotein som är nödvändigt för ett av stegen i tyrosinnedbrytningen i kroppen. [ 4 ], [ 5 ]
Således är alkaptonuri en defekt i enzymet homogentisat-1,2-dioxygenas, eller mer exakt, resultatet av dess genetiskt betingade brist eller fullständiga frånvaro. [ 6 ]
Eftersom alkaptonuri är en medfödd enzymbrist ärvs det autosomalt recessivt, det vill säga att för att alkaptonuri ska förekomma hos barn måste båda föräldrarna ha en modifierad gen för enzymet, eftersom var och en av dem endast överför en kopia av genen av de två tillgängliga till barnet.
Enligt de senaste uppgifterna finns det mer än tvåhundra varianter av modifiering av HGD-genen, och missense-mutationer, translokation och splitsning observeras oftast.
Riskfaktorer
Den enda riskfaktorn för att utveckla denna medfödda enzymopati är dess närvaro i familjehistorien och nedärvning av två modifierade kopior av HGD-genen, om föräldrarna inte uppvisar alkaptonuri (risken att överföra anomalin är 25 %), eller om en av föräldrarna har denna sjukdom. [ 7 ]
Patogenes
Tyrosin spelar en nyckelroll i syntesen av proteiner, produktionen av kromoproteiner – hudpigmentet melanin, samt sköldkörtelhormoner och katekolaminneurotransmittorer.
Mekanismen för att reglera mängden tyrosin i celler är mycket komplex, och kroppen normaliserar dess överskottsinnehåll genom att bryta ner det. Processen för tyrosinkatabolism, liksom alla aromatiska aminosyror, är flerstegs och sker i flera steg. Varje steg i den metaboliska nedbrytningen av tyrosin sker med deltagande av ett specifikt enzym och bildandet av en mellanliggande förening.
Så först bryts aminosyran ner till para-hydroxifenylpyruvat, som omvandlas till alkapton – 2,5-dihydroxifenylättiksyra eller homogentisinsyra. Sedan borde alkaptonen omvandlas till maleättiksyra, men detta händer inte. [ 8 ]
Och patogenesen för alkaptonuri består i att de biokemiska reaktionerna av tyrosinkatabolism upphör i stadiet av bildning av homogentisinsyra: det behövs helt enkelt inget enzym för att bryta ner det – homogentisatoxidas.
Homogentisinsyra används inte av kroppen och kan ackumuleras vid utsöndring via njurarna. Dessutom oxideras den till bensokinoacetat (bensokinonättiksyra), som genom att binda till molekyler i vävnader och kroppsvätskor bildar biopolymerföreningar färgade som melanin.
Ansamlingen av dessa mellanprodukter i vävnaden leder till en störning av broskvävnadens kollagenstruktur, vilket minskar dess elasticitet – med uppkomsten av många kliniska tecken på alkaptonuri och utveckling av komplikationer.
Symtom alkaptonuri
Alkaptonuri hos nyfödda och spädbarn kännetecknas av att urinen blir mörkare. När urinen på blöjor, leksaker och underkläder utsätts för luft blir den mörkbrun; detta beror på ansamling och frisättning av homogentisinsyra, som oxideras till bensokinoacetat. [ 9 ]
I avsaknad av andra symtom upptäcks alkaptonuri hos små barn ofta inte i tid, eftersom urinen kan mörkna efter flera timmars urinering. Enligt vissa uppgifter identifieras endast en femtedel av barn under 12 månader som föds med denna enzymbrist kliniskt. Därför är det mycket viktigt för föräldrar att vara uppmärksamma på att ta hand om sina spädbarn.
Dessutom inkluderar tidiga tecken pigmentering (blågrå färg) av ögonens senhinna och brosket i öron och näsa, vilket ofta kallas ochronos.[ 10 ]
Med tiden uppstår andra symtom:
- svår pigmentering av huden på kindbenen, armhålorna och könsorganen;
- fläckar på kläder vid kontakt med svettiga områden på kroppen;
- attacker av allmän svaghet;
- hes röst.
Man bör komma ihåg att alkaptonuri och ochronos, som nämnts ovan, är synonyma namn för samma sjukdom i tyrosinkatabolismen.
Lönnsirapsurinsjukdom och alkaptonuri. Medfödd lönnsirapsurinsjukdom eller leucinos är också en metabolisk sjukdom, har samma arvsmönster, och även mutationer förekommer på samma kromosom, men påverkar genen som kodar för enzymkomplexet av grenad α-ketosyradehydrogenas. På grund av detta kan kroppen inte bryta ner vissa komponenter i proteiner, i synnerhet aminosyrorna leucin, isoleucin och valin. Vid denna sjukdom har urin (och öronvax) en söt lukt; dessutom inkluderar den kliniska bilden av denna typ av organisk acidemi hypopigmentering, fluktuationer i blodtryck, kramper, kräkningar och diarré, en sänkning av blodsockernivåerna, ketoacidos, hallucinationer, etc. Dödligheten hos barn är ganska hög; hos vuxna kan koma och död uppstå utan behandling på grund av hjärnödem.
Albinism och alkaptonuri är "förenade" endast av tyrosin. Albinism, inklusive okulokutan, orsakas av genetiska mutationer som påverkar produktionen av pigmentet melanin. Medfödda förändringar noteras i TYR-genen på kromosom 11 (11q14.3), som kodar för tyrosinas, ett kopparinnehållande melanosomenzym som är nödvändigt för bildandet av hudpigment baserat på tyrosinmetabolismprodukter. Denna sjukdom är mycket vanligare än alkaptonuri.
Komplikationer och konsekvenser
Orsakad av verkan av intermediära metaboliter av tyrosin – homogentissyra och bensokinonättiksyra – uppstår konsekvenserna och komplikationerna av alkaptonuri på grund av avsättning av reaktiva pigmenterade polymerer, förstörelse av kollagenfibriller och försämring av broskets tillstånd (med en minskning av deras motståndskraft mot mekanisk stress).
Med åren, i vuxen ålder, utvecklas degenerativ artrit och artros i stora leder (höft, korsbenet och knä); intervertebralutrymmena smalnar av (särskilt i ländryggen och bröstryggen) – med förkalkning och bildande av osteofyter; vävnadstätheten i de subkondrala benplattorna minskar, och de underliggande benen kan genomgå patologisk ombyggnad med bildandet av utväxter och deformation. [ 11 ]
Skador på hjärtklaffarna (aorta- och mitralisklaffarna) och kranskärlen kan observeras – med tecken på kranskärlssjukdom, såväl som stenbildning i njurarna och prostatan – på grund av samma förkalkning. [ 12 ], [ 13 ]
Diagnostik alkaptonuri
Vanligtvis baseras diagnosen av medfödda metabola störningar på studien av kroppens biologiska vätskor.
Baserat på vilka tester och reaktioner kan alkaptonuri diagnostiseras? Urinprov krävs för att detektera homogentisinsyra och bestämma dess nivå (normal – 20–30 mg per dag, förhöjd – 3–8 g). Ett urinprov undersöks med gaskromatografi eller masspektrometri med hjälp av vätskekromatografi; ett screeningtest för förekomst av järnklorid i urinen är möjligt. [ 14 ]
Det finns också en metod för snabb diagnostik – bestämning av alkapton i torkade urinfläckar på papper (genom färgintensitet).
Vid klargörande av diagnosen innebär instrumentell diagnostik (röntgen) att identifiera radiologiska tecken på artros och andra ledpatologier hos patienter.
Diagnosen bekräftas med molekylärgenetiska metoder för att diagnostisera ärftliga sjukdomar, såsom genetisk testning och DNA-sekvensering. [ 15 ]
Differentiell diagnos
Differentialdiagnos inkluderar hemokromatos och akut leversvikt hos nyfödda, melaninuri, akut intermittent porfyri, hemofagocytisk lymfohistiocytos, primär mitokondriell patologi, reumatoid artrit, ankyloserande spondylit.
Vem ska du kontakta?
Behandling alkaptonuri
Den huvudsakliga behandlingen för alkaptonuri är oral administrering av stora doser (minst 1000 mg per dag) askorbinsyra. Hos barn ökar detta utsöndringen av homogentisinsyra i urinen, och hos vuxna minskar det urinhalten av dess derivat, bensokinonättiksyra, och saktar ner dess bindning till bindvävsstrukturer i lederna och kollagen. [ 16 ]
Västeuropeiska kliniker testar läkemedlet Nitisinone (Orfalin), ett läkemedel från gruppen metaboliter som hämmar det andra steget av tyrosinkatabolism: omvandlingen av para-hydroxifenylpyruvat till homogentisinsyra. Användningen av detta farmakologiska medel leder dock till ansamling av tyrosin och kan orsaka allvarliga biverkningar, inklusive hornhinnegrumling och fotofobi, näsblod och magblödning, leversvikt, förändringar i blodet etc. I USA är Nitisinone dock godkänt av FDA för behandling av typ I- tyrosinemi. [17 ], [ 18 ]
Därför utförs fysioterapibehandling – träningsterapi för att öka muskelstyrkan och förbättra ledrörligheten, balneoterapi och peloidbehandling för att begränsa smärta – för ledproblem orsakade av alkaptonuri.
Även om tyrosin inte bara tillförs från maten utan även produceras i kroppen, rekommenderas patienter med alkaptonuri att följa en proteinfattig kost och begränsa konsumtionen av livsmedel rika på tyrosin, främst nötkött och fläsk, mejeriprodukter (särskilt ostar), baljväxter, nötter och frön.
Förebyggande
Att förebygga genmutationer är omöjligt, men för att förhindra att barn med hög risk för medfödda sjukdomar föds finns medicinsk genetisk rådgivning, vilket är nödvändigt före en planerad graviditet för de par vars familjehistoria inkluderar ärftliga sjukdomar. [ 19 ]
Prognos
Dödlig utgång av alkaptonuri är mycket sällsynt, och dödsfallet kan orsakas av allvarliga komplikationer som involverar hjärtat och njurarna. Så den totala förväntade livslängden för personer med alkaptonuri är god.
Men livskvaliteten minskar på grund av intensiv smärta i leder eller ryggrad med betydande begränsning av rörligheten, ofta progressiv.