Medicinsk expert av artikeln

Nya publikationer
Spinal muskelatrofi
Senast recenserade: 07.06.2024

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
Spinal muskelatrofi är inte en enda nosologisk enhet, utan en hel grupp kliniskt och genetiskt heterogena ärftliga patologier som framkallas av de ökande processerna för degeneration av motoneuroner i de främre ryggraden. Termen omfattar olika varianter av genetiskt bestämd perifer pares och muskelatrofi till följd av degeneration av spinal motorneuroner och/eller hjärnstam. Den vanligaste orsaken till problemet är en autosomal recessiv mutation på den långa Q-axeln av den femte kromosomen. Behandlingen är ospecifik, som syftar till att förbättra troficiteten i nervvävnaden och ge palliativt stöd för att förbättra livskvaliteten. [1]
Epidemiologi
Spinal muskelatrofi förekommer i ett fall per 6 000 till 10 000 nyfödda (enligt American Journal of Medical Genetics 2002).
Förekomsten av SMN-gen exon 7-deletionsbärare är 1:50 personer.
Bulbo-spinal muskelatrofi (Kennedy-syndrom) förekommer hos ett barn i 50 000 och är den vanligaste vuxna typen av ryggmärgs amyotrofi.
Det noteras att hälften av barnen med denna sjukdom inte övervinner den tvååriga överlevnadsperioden.
Patologin ärvs enligt den autosomala recessiva principen. Oftast är varje förälder till ett sjukt barn en bärare av en kopia av den muterade genen. Eftersom mutationen kompenseras av närvaron av en andra "normal" genkopia, har föräldrarna inga manifestationer av spinal muskelatrofi. Typ 2-patologi ärver vanligtvis inte en ytterligare kopia från föräldern. Problemet uppstår på grund av ett oavsiktligt fel under bildningen av bakterieceller, eller direkt vid befruktningstillfället. Med spinal muskelatrofi av den första typen sker spontan utveckling av sjukdomen endast i 2% av fallen (i denna situation är bäraren bara en av föräldrarna). [2]
Orsaker spinal muskelatrofi
Den huvudsakliga orsaken till spinal muskelatrofi är en mutation av genen som är ansvarig för produktionen av SMN-protein lokaliserat på kromosom 5q. Denna störning orsakar vidare den gradvisa döden av motoriska nervceller i de främre hornen i ryggmärgen och hjärnstammen. Som ett resultat av dessa processer sjunker tonen i muskulaturen, atrofi av andnings-, svalg-, ansikts- och skelettmuskler. Den dominerande typen av arv av pediatriska former av ryggradsmuskulös atrofi är autosomal recessiv, vilket innebär samtidigt transport av defekta gener av båda föräldrarna. När det gäller patologi av typ IV (vuxenform) finns det en länk till X-kromosomen, så endast män påverkas.
Utvecklingen av ryggmuskelatrofi är baserad på de ökande processerna för degeneration och död av motoriska neuroner i ryggradens främre horn, skador på hjärnstamkärnorna. Patologiska förändringar är mest intensiva i zonerna i cervikal och ländryggen förtjockning. Det cellulära antalet reduceras till ett minimum, ersättning med bindväv inträffar, vilket beror på misslyckandet i celldödsprogrammet - den så kallade apoptosen. Förändringen påverkar strukturerna i motorkärnorna i kranialnerver, främre rötter, motoriska nerver. Det finns en klinik med neurogen fascular atrofi. Med en långvarig sjukdomskurs i ett sent skede av överväxt av bindvävnader inträffar.
Utseendet på motsvarande kliniska bild är associerad med en brist på SMN-proteinet, vilket påverkar den framgångsrika funktionen hos motornervceller i de främre ryggraden. Proteinbrist som en av länkarna i utvecklingen av ryggradens muskelatrofi upptäcktes i slutet av XX-talet. Mot bakgrund av motoneuronskador försämras innervation av skelettmuskler (främst proximala sektioner). [3]
Riskfaktorer
Mångfalden av kliniska former av ryggradsmuskulös atrofi 5Q förklaras av närvaron av vissa modifierande faktorer som kan delas in i två kategorier: de som påverkar och de som inte påverkar SMN-proteinpoängen.
- För närvarande anses SMN2-genen vara den grundläggande faktorn i utvecklingen av spinal muskelatrofi: ju fler kopior av SMN2-genen, desto lägre intensiteten hos sjukdomssymtomen. Den andra faktorn, som är direkt relaterad till den centromera kopian av SMN-genen, är en 1-nukleotidsubstitution c.859g & gt; C i exon 7 i SMN2-genen, vilket leder till bildandet av en ny förstärkare-bindande skarvplats: resultatet är inkluderingen av exon 7 i transkriptet från SMN2-genen. Denna variation är förknippad med en ökning av blodnivån för SMN-protein i full längd hos patienter med spinal amyotrofi av den andra eller tredje typen.
Andra faktorer som påverkar antalet SMN:
- Skärmreglerande faktorer (TRA2p - inducerar exon-hoppning av exon 7, SF2/ASF - ökar exon 7-inkludering, hnrnpa1 - undertrycker exon 7 inkludering av SMN2-genen).
- Transkriptionsregleringsfaktorer (CREB1 - ökar SMN-transkription, STAT3 - gynnar axontillväxt, IRF1 - ökar SMN-antalet, Prl - ökar livslängden i svåra steg).
- MRNA-stabiliserande faktorer (U1A-reducerar SMN, HUR/P38).
- Faktorer som påverkar modifiering efter translationell (RCA - undertrycker SMN-nedbrytning, GSK3 - ökar överlevnaden).
- Exogena faktorer (svält, hypoxi, oxidativ stress).
Effekterna av ovanstående faktorer bestämdes främst in vitro.
- Faktorer som inte är associerade med SMN-genen-i synnerhet proteiner som optimerar endocytos vid synapser (laminin 3, Coronin, Neurocalcin Delta, Calcium-neurin-liknande protein).
Ytterligare uppmärksamhet ägnas åt DNA-metylering, den mest stabila modifieringen som påverkar arten av genuttryck. Metyleringen av en grupp gener som eventuellt är involverade i patogenetiska processer visade sig vara korrelerade med svårighetsgraden av spinal muskelatrofi. [4]
Patogenes
Spinal muskelatrofi är en genetisk patologi för vilken någon av de typer av arv - både autosomal dominerande och autosomal recessiv eller X-kopplad - är inneboende. Oftast pratar vi om autosomal recessiv patologi i tidig barndom. Ansvaret för bildandet av sådan ryggrad amyotrofi är SMN-genen, lokaliserad på lokuset 5q13. Radering av exon 7 i SMN-genen resulterar i patologi med möjlig involvering av de närliggande generna p44 och NAIP.
SNM-genomet kodar för ett protein som inkluderar 294 aminosyror och har en mM av ~ 38 kDa. Proteinet har följande funktioner:
- Är en del av RNA-proteinkomplexet;
- Deltar i bildandet av spliceosomstället som katalyserar pre-RNA-skarvning;
- Involverad i processer som kontrollerar proteinproduktion och proteinisoformer;
- Tillhandahåller axonal transport av mRNA;
- Gynnar nervcelltillväxt och ger neuromuskulär kommunikation.
Ett par typer av SMN-gener är kända:
- Telomer SMNT (SMN1);
- Centromer SMNC (SMN2).
De allra flesta fall av spinal muskelatrofi beror på förändringar i SMN1-genen.
Kennedy spinal muskelatrofi har en koppling till XQ12-lokuset som innehåller NR3C3-genen, som kodar ett androgenreceptorprotein. Den har en X-kopplad arvvariant. När antalet CAG-upprepningar i en genexon ökar utvecklas patologin.
Undertryckning av SNM-proteinproduktion åtföljs av följande förändringar:
- På grund av nedsatt axonkoordination inträffar överdriven förgrening av axoner;
- Tillväxten av axoner bromsar och deras storlek minskar;
- Det finns felaktig kluster av kalciumkanaler i tillväxtkonen;
- Oregelbundna presympatiska terminaler för motorens nervcellsaxoner bildas.
Ryggmärgen börjar aktivt förlora motoriska neuroner i de främre hornen, som står för utvecklingen av atrofi i de proximala lemmusklerna. [5]
Symtom spinal muskelatrofi
Symtomatologi av ryggradsmuskulös atrofi Werdnig-hoffman debuterar oftast under den nyfödda perioden och upp till sex månader, som manifesteras av syndromet av ett "trögt" barn. Det klockformade bröstet, intensiv hypotoni, brist på reflexer, muskel ryckning av tungan och andningsbesvär märks. Sjuka spädbarn dör oftare innan de fyller två år: dödligt resultat beror på ökande andningsfel mot bakgrund av vidhäftningen av infektiösa processer.
Den mellanliggande formen av spinal muskelatrofi av den andra typen detekteras från sex månader. Förutom syndromet för ett "trögt" barn finns det lågt blodtryck, brist på reflexer, andningsstörningar och tunga ryckningar. Även om barn kan sitta upp utvecklas flera kontraktioner i de stora lederna.
Kugelberg-Wielander spinal muskelatrofi börjar också i tidig barndom, med barn som kan röra sig självständigt. Det försvagas av iliac-, quadriceps- och adduktormusklerna, lågt blodtryck, minskade reflexer och tunga ryckningar. Många patienter förlorar förmågan att flytta (gå) oberoende under åren.
Spinal muskelatrofi typ 4 börjar vid en äldre ålder. Det kännetecknas av långsam progression och relativt godartad prognos. [6]
Kennedy Atrophy manifesterar sig oftast i medelåldern (i allmänhet kan debutera hos patienter 15-60 år). Symtomatologi inkluderar muskelsår och svaghet, gynekomasti, distal svaghet, slöhet, tunga ryckning och atrofi. Tecken på bulbar dysfunktion finns:
- Svårigheter att svälja;
- Strävan;
- Försvagning av de masticatoriska musklerna;
- Dysartri;
- Postural och motoriska skakningar i händerna.
Första tecken på androgenbrist:
- Gynekomasti (hos cirka 60% av patienterna), ofta asymmetriska;
- Försämring av sexuell funktion (oligospermia, testikelatrofi, erektil dysfunktion).
Första tecken
Spinal amyotrofi manifesteras av svaghet i muskler och allmän impotens. Alla sensoriska och intellektuella förmågor påverkas inte.
Viktiga index för neuromuskulär patologi:
- Muskulatur "lat", försvagad, musklet och slapphet i musklerna noteras;
- Muskeltonen är låg, senreflexer är minimerade eller frånvarande;
- Normala eller frånvarande plantarreflexer;
- Korta ryckningar av enskilda muskelgrupper noteras (kan ses under huden, på tungan);
- Det finns tecken på muskelatrofi.
Werdnig-Hoffman-syndrom manifesteras av uttalad hypotoni av musklerna, allmänt slöhet, oförmåga att hålla huvudet, vända och sitta upp. När man försöker stödja barnet i buken i ett hängande tillstånd verkar kroppen "sjunka". Hosta, svälja och suga reflex är otillfredsställande, mat kommer ofta in i luftvägarna, andningen är problematisk. Det kan vara ledförvrängning i samband med intrauterin hypotoni. Anamnestic information som samlats in under graviditeten indikerar ofta låg fosteraktivitet.
Grundläggande tecken på spinal muskelatrofi typ I:
- Allvarlig retardering i motorutvecklingen;
- Snabb början av gemensamma kontrakturer och thorax krökning;
- Ökande störningar och bulbar-störningar, problem med sväljning (både mat och saliv) och expektion av sputum;
- Ökad risk för ambitionsinflammation;
- Infektion, progressivt andningsfel.
Spinal muskelatrofi typ II manifesteras av en tydlig hämning av motorisk utveckling. Även om många patienter kan sitta utan hjälp, och ibland till och med krypa och stå, förloras dessa förmågor ofta över tid. Fingerskakningar, muskler och leder (ben) snedvridningar och andningsproblem noteras. Möjlig kalvpseudohypertrofi.
De viktigaste funktionerna i typ II-patologi:
- Utvecklingsförseningar, inklusive att stoppa och vända utvecklingen av redan förvärvade färdigheter och förmågor;
- Ökande svaghet i de interkostala musklerna;
- Ytlighet av membran andning, försvagad hostreflex, gradvis försämring av andningsfel;
- Krökning av bröstkorgen och ryggraden, kontrakturer.
I Kugelberg-Wielander-syndromet är manifestationerna mildare och utvecklas långsamt. Patienten kan röra sig, men det finns problem med jogging eller klättring trappor. Försenade symtom inkluderar ofta svårigheter att svälja och tugga.
Spinal muskelatrofi typ IV avslöjar sig redan i äldre (vuxna) ålder och kännetecknas av den mest "milda" och gynnsamma kursen. Huvudtecknen: Gradvis förlust av förmågan att flytta. [7]
Formulär
Spinal muskelatrofi är en del av en grupp av ärftliga patologier som kännetecknas av degenerativa förändringar, döden av motoriska nervceller i de främre ryggraden och ofta de motoriska kärnorna i hjärnstammen. Processen kan göra sig känd under olika livsperioder, den kliniska bilden är inte alltid densamma. Typerna av arv och kurs kan också skilja sig åt.
Pediatrisk spinal muskelatrofi beskrevs först redan i slutet av 1800-talet. Runt mitten av 1900-talet identifierades de viktigaste formerna av sjukdomen:
- Medfödd (manifesterar sig nästan omedelbart efter spädbarnets födelse);
- Tidig infantil form (inträffar mot bakgrund av tidigare normal utveckling av barnet);
- Sen infantil form (avslöjar sig från och med 2 år och äldre).
Vissa specialister kombinerar de andra och tredje formerna till en pediatrisk typ av ryggradsamyotrofi.
Det är allmänt accepterat att dela upp patologi i pediatrisk och vuxen. Spinal muskelatrofi hos barn klassificeras i tidigt (med en debut under de första månaderna efter barnets födelse), sent och ungdomar (tonåringar eller ungdomar). De syndrom som oftast är involverade är:
- Werdnig-Hoffman atrofi;
- Kugelberg-Wielander-formen;
- Kronisk infantil spinal muskelatrofi;
- Vialetto-van Lare-syndrom (bulbospinal typ med frånvaro av hörsel);
- Fazio-Londe syndrom.
Vuxen spinal muskelatrofi debuterar över 16 år och fram till ungefär 60 års ålder, kännetecknad av en relativt godartad klinik och prognos. Vuxna patologier inkluderar:
- Kennedys bulbospinal atrofi;
- Scapuloperoneal atrofi;
- Ansiktsvarv-axel- och oculo-faryngeala former;
- Distal ryggradsatrofi;
- Monomelisk spinal atrofi.
Separat separat isolerad och kombinerad ryggradsatrofi. Isolerad patologi kännetecknas av övervägande av skador på spinalmotorneuroner (som ofta är det enda tecknet på problemet). Kombinerad patologi är sällsynt och representerar ett komplex av neurologiska och somatiska störningar. Det finns beskrivningar av fall av kombinerat syndrom med medfödda koronära missbildningar, brist på hörselfunktion, oligofreni, cerebellär hypoplasi.
Spinal muskelatrofi hos äldre representeras oftast av Kennedy bulbospinal amyotrofi. Denna patologi ärftas recessivt X-länkad. Sjukdomens gång är långsam, relativt godartad. Det börjar med atrofi av den proximala muskulaturen i de nedre extremiteterna. Möjlig skakning av händerna, huvudet. Samtidigt upptäcks också endokrina problem: testikelatrofi, gynekomasti, diabetes mellitus. Trots detta, hos vuxna, fortsätter patologin i en mildare form än hos barn.
En variant av spinal muskelatrofi. |
Debuten för patologin |
Detekterbart problem |
Dödsålder |
Karakteristisk symptomatologi |
Spinal muskelatrofi typ 1 (annat namn Verding-hoffman spinal muskelatrofi) |
Från födseln till sex månader |
Barnet kan inte sitta upp |
Upp till två år |
Allvarlig muskelsvaghet, hypotoni, problem med att hålla upp huvudet, nedsatt gråt och hosta, svälja och salivproblem, utveckling av andningsfel och aspiration lunginflammation |
Spinal muskelatrofi typ 2 |
Sex månader till ett och ett halvt år |
Barnet kan inte stå |
Mer än två år |
Motorfördröjning, viktbrist, hosta svaghet, handskakningar, ryggrad, kontrakturer |
Spinal muskelatrofi typ 3 (annat namn Kugelberg-Welander spinal muskelatrofi) |
Efter ett och ett halvt år. |
Kan initialt stå och gå, men i en viss ålder kan denna förmåga gå förlorad |
I vuxen ålder. |
Försvagade muskler, kontraktioner, gemensam hypermobilitet |
Spinal muskelatrofi typ 4. |
Tonåren eller vuxen ålder |
Kan initialt stå och gå, men i en viss ålder kan denna förmåga gå förlorad |
I vuxen ålder. |
Ökande proximal muskelsvaghet, minskade senreflexer, muskel ryckning (fascikulationer) |
Om distal ryggradsatrofi sägs i fallet med lesioner av motoriska nervceller i ryggmärgen, som innerverar den nedre delen av kroppen. Karakteristiska tecken på sådan patologi är:
- Atrofi av lårmusklerna;
- Svaghet i knäna, fotförlängare och höftadduktormuskler.
Ingen förändring i senreflexer.
Distal spinal muskelatrofi representeras av två alleliska variationer med en överlappande fenotyp:
- Scapulo-perineal spinal muskelatrofi;
- Ärftlig motorsensorisk neuropati av Charcot-Marie-Tooth typ 2C.
Proximal spinal muskelatrofi 5Q kännetecknas av ökande symptomatologi för slapp förlamning och muskelatrofi, vilket beror på degenerativa förändringar i alfa-motorneuronerna i de främre ryggraden. Medfödd sjukdom med asfyxi efter födseln är den allvarligaste formen: Från det ögonblick som barnet föds, är motorisk aktivitet praktiskt taget frånvarande, det finns kontraktioner, sväljning och andningsproblem. I de flesta fall dör ett sådant barn.
Komplikationer och konsekvenser
Ytterligare progression av ryggradens amyotrofi leder till svaghet och minskning av muskelmassan i lemmarna (särskilt ben). Barnet har initialt inte eller förlorar gradvis förvärvade färdigheter - det vill säga förlorar förmågan att gå, sitta utan stöd. Motorisk aktivitet i de övre extremiteterna minskar, lederna blir styva, över tidskontrakturerna är fästa och ryggraden blir krökt.
För att bevara motoriska förmågor så länge som möjligt och förhindra utveckling av komplikationer rekommenderas det:
- Öva korrekt kroppsställning (anti-gravity position), både i sängen och när du sitter, går osv.
- Regelbunden fysioterapi, stretchövningar, massage, fysioterapi, oavsett typ av muskelatrofi av ryggraden;
- Använd specialbäddar, stolar (rullstolar), madrasser och kuddar;
- Välj och använd stödjande ortotik, korsetter;
- Öva hydroterapi och kinesioterapi, som har en gynnsam effekt på andnings-, muskel- och matsmältningsapparaten, nervös och hjärt-kärlsystem;
- Utför regelbundna diagnostiska kontroller, inklusive kliniska tester, ryggrads- och bäckenradiografer;
- Konsultera systematiskt med en fysioterapeut och ortoped med erfarenhet av att arbeta med liknande patienter;
- Justera korsetter, ortoser, ortopediska enheter, rullstolar etc. beroende på dynamiken.
Vårdgivare av en patient med spinal muskelatrofi bör bekanta sig:
- Med grunderna för säkert beteende, fysioterapi, massage, fysioterapi;
- Med reglerna för att upprätthålla oberoende aktivitet hos patienten, användning av ortopediska apparater;
- Med reglerna för vård, hygien.
Spinal amyotrofi kompliceras ofta av nedsatt tuggning, sväljning och ledning av mat, vilket hotar aspiration och utvecklingen av aspirationsinflammation i lungorna eller hindring av luftvägarna, som är mest karakteristisk för patologin för den första typen. Att svälja problem bevisas av symtom som betydande och ihållande förlängning av ätningsperioden, motvilja mot att äta, mat som faller ut ur munnen, regelbunden gagging och förvärrande viktminskning.
Störningar i matsmältningsmotiliteten avslöjar sig förstoppning, svag peristaltik, långvarig mat i magen (gastrisk stas), utvecklingen av gastroesofageal återflöde. För att förhindra sådana komplikationer är det nödvändigt:
- Övervaka patientens korrekta läge när du äter;
- Använd vid behov ett gastriskt rör eller gastrostomi för att säkerställa adekvat vätska och näringsintag och minska risken för aspiration;
- Följ reglerna för mat- och dryckförberedelser, titta på deras konsistens och frekvensen av måltider;
- Beroende på läkarens recept, använd medicinering, massage, fysioterapi etc.
En av de mest allvarliga komplikationerna av ryggradens amyotrofi är andningssystemdysfunktion förknippat med svaghet i andningsmusklerna. Andningsstörningar kan vara dödliga, både hos spädbarn med typ 1-patologi och hos ungdomar och vuxna patienter med typ 2 eller 3 sjukdom. De viktigaste problemen är följande:
- Hosta reflex är störd, det finns problem med expectoration av sputum från luftvägarna;
- Ökande underskott i luftvolymen som kommer in i lungorna, nedsatt utsöndring av koldioxid från lungorna;
- Snedvrider bröstet, komprimerar och deformeras lungorna;
- Infektionsprocesser i form av bronkopneumoni.
För att förhindra sådana komplikationer rekommenderas ofta patienter att utföra andningsövningar med en Ambu-väska. [9]
Diagnostik spinal muskelatrofi
Hos patienter med misstänkt ryggradsamyotrofi är undersökningar som dessa av diagnostiskt värde:
- Blodkemi;
- Genetisk DNA-analys;
- Elektronuromyografi.
Bland ytterligare metoder är det möjligt att utse en biopsi av muskelfibrer, ultraljud och resonansavbildning av muskulaturen och hjärnan.
Blodtester kan indikera att kreatinfosfokinas är fysiologiskt normalt, men i vissa fall kan det höjas till cirka 2,5 gånger.
Elektronuromyogrammet avslöjar förändringar på grund av förlusten av motoriska spinalneuroner. Detta detekteras genom en minskning av amplituden av störningskurvan, förekomsten av spontana aktiva potentialer, som är fibrillationer och fascioculations som bildar en specifik "frekvensrytm". Hastigheten för impulssignal som passerar genom perifera motorfibrer är normal eller minskas på grund av sekundära denervationsstörningar. [10]
Instrumental diagnos representeras ofta också av ultraljud eller MR-muskulatur, som möjliggör detektering av muskelbyte med fettvävnad. MRI avslöjar ett typiskt patologiskt processmönster unikt för ryggmuskulär atrofi. Detta är emellertid endast möjligt i de sena stadierna av lesionen.
Under morfologisk analys av muskelbiopsi hos patienter bestäms en ospecifik bild i form av buntatrofi och gruppering av muskelfibrer. Det överväldigande antalet drabbade muskelfibrer tillhör typ 1, immunohistologiska och kemiska egenskaper ligger inom normala gränser. Den ultrastrukturella bilden är ospecifik.
Den viktigaste diagnostiska proceduren för misstänkt spinal muskelatrofi är testning som kan upptäcka SMN-genmutationen. Genom direkt DNA-analys är det möjligt att upptäcka närvaron eller frånvaron av de sjunde och åttonde exonerna av SMNC- och SMNT-generna. Den mest informativa metoden är kvantitativ analys, som kan bestämma genkopieringsnumret och belysa formen av ryggradsmuskulös atrofi. Den kvantitativa metoden är också viktig för att bedöma patientens status. Det är en nödvändig åtgärd som genomförs för ytterligare medicinsk och genetisk familjerådgivning.
Ytterligare diagnostiska tester utförs endast efter att ett negativt resultat av SMN-gendeletion erhålls. Om detekteringen av punktmutationer krävs kan direkt automatiserad sekvensering av SMNT-genen användas.
Differentiell diagnos
Differentialdiagnosen utförs med patologiska processer som avslöjar symptomkomplexet för "trög patient", med medfödda muskeldystrofier, strukturell eller mitokondriell myopati. I synnerhet bör närvaron av sådana patologier uteslutas:
- Motorneuronsjukdom;
- Primär lateral myoskleros;
- Muskeldystrofi;
- Medfödda myopatier;
- Sjukdomar förknippade med glykogenansamling;
- Polio;
- Autoimmun myasthenia gravis.
Den diagnostiska algoritmen utvecklas beroende på särdragen i symptomatologin hos ett visst barn. Således används en speciell klassificering av patienter, beroende på funktionell status (Europrotocol Treat-NMD):
- Det går inte att sitta upp utan stöd (sängliggande).
- Kunna sitta men inte kunna gå (stillasittande).
- Kunna röra sig självständigt (promenader).
Följande diagnostisk algoritm rekommenderas för patienter i den första gruppen:
- Fysisk undersökning (upptäckt av bröstkrökning, bedömning av andnings- och hostfunktion och hudtillstånd);
- Hjärt- och andningsövervakning, polysomnografi och identifiering av symtom på underskott av lungventilation;
- Pulsoximetri för att bestämma graden av syresättning;
- Bedömning av frekvensen av infektionsinflammatoriska patologier och antibiotikakurser under den extrema sexmånadersperioden;
- Bröströntgenstrålar med upprepade dynamikstudier;
- Bedömning av sväljningsfunktionen.
För patienter i den andra gruppen gäller följande algoritm:
- Fysisk undersökning;
- Hjärt- och andningsövervakning, polysomnografi för att upptäcka underskott av lungventilation;
- Pulsoximetri;
- Bedömning av frekvensen av infektionsinflammatoriska processer och antibiotikakurser under den extrema sexmånadersperioden;
- Undersökning av ryggraden, röntgenstrålarna i ryggraden, bedömning av graden av krökning.
Patienter i den tredje gruppen indikeras för sådana studier:
- Fysisk undersökning;
- Andningsfunktionstest (inkluderar spirometri, beräkning av lungvolym, bedömning av andningsmuskelfunktion);
- För att ta reda på frekvensen av infektionsinflammatoriska patologier och antibiotikakurser under den extrema årsperioden.
Utövandet av differentiell diagnos kan kompliceras av likheten mellan SMN1- och SMN2-gener. För att undvika fel rekommenderas det att använda MLPA-metoden, som gör det möjligt att upptäcka kopieringsnumret för exon 7 i SMN1-genen.
I de flesta fall av spinal muskelatrofi finns det en homozygot borttagning av exon 7 och/eller 8 i SMN1-genen. Men andra gener (Atp7a, DCTN1, Uba1, BSCl2, EXOSC3, GARS, etc.) kan också vara "skyldiga", vilket bör uppmärksammas om SMN1-testet är negativt.
Biomaterialet för studien kan vara perifert blod eller fosterblod, kartor med torr blod. Diagnos är obligatorisk:
- I närvaro av en förvärrad historia av ryggradsmuskulös atrofi;
- När misstänkta symtom upptäcks, oavsett ärftlig historia.
Dessutom rekommenderas forskning också för alla par som är ansvariga för att planera en graviditet.
Vem ska du kontakta?
Behandling spinal muskelatrofi
Patienter med spinal muskelatrofi behöver omfattande behandling som inkluderar:
- Omsorg, hjälp, stöd;
- Dietmat;
- Läkemedelsbehandling;
- Icke-mediceringsrehabiliteringsåtgärder, inklusive kinesioterapi och fysioterapi.
En terapeutisk behandling som involverar en polymodal effekt på alla kroppssystem, inte bara muskuloskeletalsystemet, är standard.
Tyvärr är det omöjligt att radikalt bota ryggmuskelatrofi. Men det är ofta möjligt att förbättra patientens livskvalitet genom kompetent användning av aminosyror och multivitaminkomplex, neurotrofiska medel, kalciumkanalblockerare, vasodilatatorer, kardiotrofiska och cytostatiska läkemedel, proteasinhibitorer, steroidala läkemedel, antioxidanter, immunoglobuliner och immunoschef och så på. Det har bevisats experimentellt att behandling med stamceller, neurobeskyttande föreningar och muskelstärkande molekyler kan leda till oförutsägbara systemiska störningar. Samtidigt har positiv dynamik efter tillämpningen av sådan behandling hittills bevisats.
Eftersom problemet orsakas av en brist på normalt SMN-protein kan patienter förbättras genom att öka SMN-proteinnivåerna med 25% eller mer. Av denna anledning undersöks läkemedel som kan aktivera produktionen av detta protein aktivt, inklusive gabapentin, riluzol, hydroxyurea, albuterol, valproinsyra och natriumfenylbutyrat.
Modern medicin erbjuder också kirurgisk behandling för spinal muskelatrofi. Den består av kirurgisk inriktning av ryggraden - korrigering av neuromuskulär krökning. Kirurger utför fixering av flernivåer av ryggraden med speciella konstruktioner. Sacrum, bäcken och ryggkotor i de övre bröstkotorna eller andra ryggkotor används som stödpunkter. Operationen hjälper till att anpassa spinalkolonnen, jämnt fördela belastningen på den, eliminera obehag när man ändrar kroppens position, undviker negativa effekter på inre organ (inklusive lungorna).
Mediciner
För närvarande finns det ingen etiologisk behandling för spinal muskelatrofi: vetenskaplig medicin fortsätter att arbeta med denna uppgift. Tidigare har forskare redan lyckats isolera läkemedel som kan förbättra produktionen av mRNA från SMN2-genen. Men storskaliga internationella kliniska prövningar som involverar personer med ryggradsmuskulös atrofi har ännu inte genomförts.
De flesta av läkemedlen som ingår i standardbehandlingsregimen har en allmän handlingsprincip med relativt låga bevis på effektivitet.
L-karnitin |
En naturligt förekommande aminosyra, en "relativ" av B-gruppens vitaminer. Det produceras i kroppen, finns i levern och tvärgående strierade muskler, tillhör ett antal vitaminliknande ämnen. Deltar i metaboliska processer, stöder COA-aktivitet, används för att normalisera metabolism. Den har anabolisk, antityreos, antihypoxisk förmåga, stimulerar lipidmetabolism och vävnadsreparation, optimerar aptiten. L-karnitin förskrivs till en mängd av cirka 1 tusen mg per dag. Behandlingsförloppet kan pågå i upp till 2 månader. |
Coenzyme Q10 (ubiquinon) |
En koenzymbensokinongrupp som innehåller ett antal isoprenylgrupper. Dessa är fettlösliga koenzymer, främst närvarande i mitokondrier av eukaryota cellulära strukturer. Ubiquinon ingår i elektrontransportkedjan, deltar i oxidativ fosforylering. Den största närvaron av ämnet finns i energirika organ - i synnerhet i levern och hjärtat. Koenzym Q10 har bland annat antioxidantegenskaper, kan återställa antioxidantkapaciteten hos alfa-tokoferol. Vanligtvis föreskrivs från 30 till 90 mg av läkemedlet per dag, en två månaders kurs. |
Cerebrolysin |
Ett nootropiskt läkemedel med neurotrofiska egenskaper. Det används ofta i terapeutiska regimer för behandling av neurologiska patologier, inklusive vaskulär demens, stroke. Den aktiva fraktionen inkluderar peptider med en begränsande molekylvikt på 10 tusen daltoner. Läkemedlet administreras som en intravenös injektion av 1-2 ml. Behandlingsförloppet består av 10-15 injektioner. |
Aktovin |
Läkemedlets sammansättning representeras av peptider med låg molekylvikt och aminosyrorivat. Actovegin är ett hemoderivativ: den isoleras av dialys med ultrafiltrering. Tack vare användningen av läkemedlet ökas absorptionen och användningen av syre, energimetabolismen accelereras. Läkemedlet används i form av intravenösa injektioner av 1-2 ml, kursen kräver 10-15 injektioner. |
Solkosl |
It is a deproteinized hemodialysate capable of optimizing pre-cellular oxygen and glucose transport, enhancing intracellular ATP production, stimulating regenerative tissue reactions, activating fibroblast proliferation and collagen production in vascular walls. Behandlingsförloppet består av 10-15 intra-muskulära injektioner av läkemedlet (1-2 ml dagligen). |
Neuromultivit (vitamin B-komplex) |
Multivitamin, aktivt används i bristen på vitamin B-grupp. Det kan ofta bli en kvalitetsersättning för en kurs med injektioner av vitaminpreparat. Aktiverar metaboliska processer i hjärnan, främjar återställande av vävnader i nervsystemet, har en smärtstillande effekt. Neuromultivit tar 1-2 tabletter dagligen, en kurs på 4 eller 8 veckor. |
Vitamin E |
Ett välkänt antioxidant, fettlösligt vitamin. Det föreskrivs på kurser på 1-2 månader till 10-20 IU dagligen. |
Valproat |
De har lugnande och avkopplande aktivitet, visar antikonvulsiv förmåga, ökar nivån på GABA i CNS. Används endast för behandling av barn under ett års ålder, 10 till 20 mg per kg per dag. |
Salbutamol |
En bronkodilator, som tillhör gruppen av selektiv beta2-adrenoreceptoragonister. Regelbunden användning av läkemedlet orsakar ökad produktion av mRNA- och SMN-protein, vilket positivt påverkar den kliniska bilden av spinal muskelatrofi. Salbutamol används försiktigt, 2-4 mg fyra gånger om dagen (den maximala mängden är 32 mg per dag). |
Ett av de senaste läkemedlen som används i ryggradens muskelatrofi är Zolgensma-genoterapeutiska läkemedlet Zolgensma, vilket säkerställer aktiviteten och korrekt funktion av transducerade motoriska nervceller. Läkemedlet administreras i kombination med immunmodulerande läkemedel enligt ett speciellt protokoll och administreras en gång intravenöst, baserat på en nominell dosering av 1,1 ͯ 1014Vg/kg (den totala administreringsvolymen är bestämd beroende på patientens vikt).
Innan zolgensmabehandling startar är det obligatoriskt att bestämma nivån på antikroppar mot AAV9 med användning av en validerad diagnostisk metod, utvärdera leverfunktion (ALT, AST, total bilirubin), utföra allmän klinisk blodundersökning och troponin I-test, bestämma kreatininnivån. Om akuta och kroniska aktiva infektionsförhållanden upptäcks, skjuts administrering av läkemedlet upp till botemedel eller slutförande av återfallsfasen i den infektiösa processen.
Den vanligaste biverkningen av läkemedlet anses vara leversvikt, vilket kan vara dödligt.
Andra godkända mediciner som din läkare kan förskriva för ryggmuskelatrofi:
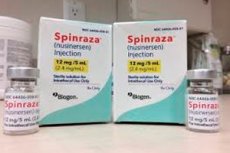
- Spinraza är en beredning av Nusinersen-natrium, en antisense oligonukleotid specifikt utformad för behandling av ryggradens amyotrofi. Det är avsett för intratekal administration genom ländryggen. Den rekommenderade dosen är 12 mg. Behandlingsregimen bestäms av den behandlande läkaren.
- Risdiplam är ett läkemedel som modifierar skarvning av mRNA-föregångaren för den motoriska nervcellens överlevnadsgen 2. Risdiplam tas oralt, en gång om dagen. Doseringen bestäms av läkaren individuellt med hänsyn till patientens ålder och vikt. Användningen av läkemedlet hos barn yngre än 2 månader är kontraindicerat. Embryofetal toxicitet för detta läkemedel noteras, så reproduktiva potentialpatienter bör vidta noggranna preventivåtgärder både under och en viss period efter behandlingen.
Fysioterapeutisk behandling för spinal muskelatrofi
Fysioterapi används som en av länkarna till komplex terapi och rehabilitering av patienter med ryggradsmuskulös atrofi. Huvudpunkterna för sådan behandling är:
- Användning av lossning med hjälp av upphängningssystem, aktiv passiv träning, användning av perkutan elektrisk stimulering av ryggmärgen;
- Andningsövningar och fysioterapi;
- Halvtimmes vertikaliseringssessioner;
- Translingual elektrostimuleringsbehandlingar (20-minuters sessioner, i kombination med övningar för att förbättra fina motoriska färdigheter);
- Manuella tekniker;
- Paraffinapplikationer på olika grupper av artikuleringar;
- Darsonval för att förbättra muskelprestanda.
Metoden för DarsonValization är baserad på effekten på vävnader med användning av en växlande högfrekvenspulström med högspänning och låg styrka. Efter en kurs för procedurer är det en ökning av muskelprestanda, förstärkning av mikrocirkulation, expansion av arterioler och kapillärer, eliminering av ischemi, förbättring av näring och syretillförsel till muskler, vilket har en positiv effekt på kursen för regenerativa och atrofiska processer.
Ett av de viktigaste problemen hos patienter med ryggraden amyotrofi är andningsmuskelsvaghet, vilket ofta leder till andningsdysfunktion och död av patienten.
I spinal amyotrofi är hela skelettmuskulaturen, inklusive det som ansvarar för andning, underpresterande. Svaghet och gradvis muskelatrofi påverkar negativt kvaliteten på andningshandlingen, leder till utveckling av komplikationer och ökande andningsfel. Därför är det nödvändigt att vidta åtgärder för att stärka musklerna, förebyggande av luftvägskomplikationer och luftvägsinfektioner. En speciell roll i detta spelar gymnastik med Ambu-väskan, som utförs i samband med fysioterapi, stretchövningar, massage. Användningen av Ambu Bag låter dig "utöka" bröstets volym och lungor. För barns aktiviteter är lämplig väska med en volym av minst en och en halv liter, utrustad med en ventil för att frigöra överdrivet tryck (för att förhindra barotrauma).
Övningar bör inte utföras på full mage. Kroppsposition - sittande, halvsittande, liggande på sidan eller ryggen (om det inte finns några problem med slem): Det är optimalt att utföra procedurerna i olika positioner varje gång. Det är viktigt att patientens rygg rätas ut. Vid behov används en korsett. Innan du startar proceduren, se till att luftvägen är fri från sputum.
Massage för spinal muskelatrofi
Massage för behandling av ryggraden amyotrofi bör vara lätt och skonsam. I områden med muskelmotstånd tillämpar allmänna effekter, inklusive tappning, och i områden med bevarad innervation använder du djup strökning (längsgående, tvärgående), knådning.
I allmänhet, utövar olika typer av massage, beroende på de enskilda egenskaperna hos sjukdomens gång, patientens ålder. Dessa kan vara:
- Knådning för att stimulera djupa sittande muskler;
- Gnuggar för att optimera blod och lymfcirkulation;
- Spotbehandling av triggerpunkter;
- Av den fiberstärkande dunken.
Det är viktigt att effekten är spridd över hela problemområdet.
Kontraindikationer för massage för ryggradsmuskulös atrofi:
- Akut inflammation, förhöjd kroppstemperatur;
- Blodstörningar, blödningstendenser;
- Purulenta processer;
- Smittsamma, svampdermatologiska sjukdomar;
- Vaskulära aneurysmer, trombangiit, endarterit, lymfadenit;
- Godartade och maligna neoplasmer.
Förloppet för varje massage för en patient med spinal muskelatrofi föreskrivs strikt individuellt. Felaktigt uppförande av förfarandet, alltför grov och felaktig påverkan kan skada patientens tillstånd.
Förebyggande
Direkt och indirekt DNA-diagnos och prenatal DNA-diagnos bedrivs nu aktivt. Detta minskar avsevärt sannolikheten för att ett sjukt barn föddes, vilket är särskilt viktigt för par som redan har upplevt födelsen av barn med ryggradsmuskulös atrofi.
Förebyggande åtgärder representerar en viktig medicinsk trend och kategoriseras i primära, sekundära och tertiära åtgärder.
Primära åtgärder syftar till att direkt förhindra påverkan av en ogynnsam faktor och förhindra utvecklingen av sjukdomen. Sådant förebyggande består i att korrigera diet och daglig regim, vilket leder en hälsosam livsstil.
Sekundär förebyggande består i eliminering av uppenbara riskfaktorer och inkluderar tidig diagnos av patologier, etablering av övervakning i dynamik, riktad behandling.
Tertiärförebyggande utförs i förhållande till en sjuk person som berövas vissa motoriska kapaciteter. I den här situationen talar vi om medicinering, psykologisk, social och arbetsrehabilitering.
Enligt information från Världshälsoorganisationen föds mer än 2% av barnen i världen med någon form av utvecklingsstörning. Samtidigt är 0,5-1% av sådana störningar av genetiskt ursprung. Förebyggande av sådana problem reduceras till medicinsk genetisk rådgivning och prenatal diagnos av kvalitet, vilket gör att minimering av riskerna för att föda ett barn med genetisk patologi.
En persons risk att få spinal muskelatrofi eller en annan genetisk sjukdom beror på generna som ärvts från hans mor och far. Tidig identifiering av ärftliga faktorer, beräkning av enskilda risker för genetiskt bestämd patologi kan kallas ett sätt att vara riktat förebyggande.
Prenatala diagnostiska åtgärder inkluderar direkta och indirekta forskningsmetoder. Ursprungligen identifieras kvinnor som kräver indirekt prenatal diagnos. Dessa kan inkludera:
- Gravida kvinnor 35 år och äldre;
- Som har haft två eller fler tidigare spontana aborter;
- Som har barn med genetiska utvecklingsfel;
- Med en ogynnsam ärftlig historia;
- Som har haft virala infektioner eller strålningsexponering (inklusive under planeringsstadiet av graviditeten).
För förebyggande ändamål används sådana metoder som ultraljud, hormonella tester (biokemisk screening). Ibland används invasiva procedurer såsom chorionbiopsi, amniocentes, placentocentes, cordocentesis. Tillförlitlig information om genetiska risker gör att du kan justera din livsstil och graviditet för att förhindra att ett sjukt barn föddes.
Vaccin mot muskelatrofi
Naturligtvis skulle alla föräldrar till barn med ryggraden amyotrofi vilja bota dem helt av sjukdomen. Det finns emellertid inget vaccin som kan utrota problemet. Även om forskning om optimering av behandling pågår.
I synnerhet godkände amerikanska forskare 2016 den unika läkemedelsspinraza (Nusinersen), som därefter godkändes för användning i europeiska länder.
Specialister undersöker problemet med att behandla spinal muskelatrofi på dessa sätt:
- Fixa eller ersätta den "fel" SMN1-genen;
- Förstärkning av funktionen hos den normala SMN2-genen;
- Skydd av motornervceller som påverkas på grund av SMN-proteinbrist;
- Skydd av muskler från atrofiska förändringar för att förhindra eller återställa förlorad funktion mot bakgrund av patologutveckling.
Genterapi involverar inriktning på den skadade genen med hjälp av virala vektorer som passerar genom blodhjärnmembranet och når lämpligt område i ryggmärgen. Sedan "infekterar" den drabbade cellen med en frisk DNA-del, som om "suturering" gendefekten. Således korrigeras funktionen hos motoriska nervceller.
En annan riktning är liten molekylterapi, vars essens är att förbättra SMN2-genens funktion. Spädbarn med diagnostiserad spinal muskelatrofi har minst en kopia av SMN2-genen. Denna riktning har aktivt undersökts av amerikanska forskare, och för närvarande genomgår flera läkemedel som syftar till att förbättra syntesen av ett komplett protein från SMN2-genen kliniska studier.
En annan väg med möjlig terapeutisk intervention är att utforska neurobeskydd för att minska motorneurondöd, öka deras anpassningsförmåga och förbättra funktionaliteten.
Den tredje riktningen involverar att skydda muskeln från atrofiska processer. Eftersom SMN-proteinbrist negativt påverkar motoriska nervceller och muskelfunktion, bör målet med denna behandling vara att skydda musklerna från atrofi, öka muskelmassan och återställa muskelfunktionen. Denna typ av terapi kommer inte att påverka den genetiska apparaten, men den kan sakta ner eller till och med blockera försämringen av spinal muskelatrofi.
Screening för spinal muskelatrofi
Nyfödd screening används alltmer i medicinsk praxis och spelar ofta en avgörande roll. Att upptäcka spinal muskelatrofi så tidigt som möjligt kan förbättra prognosen för det sjuka barnet avsevärt. Screeningdiagnos inkluderar följande punkter som beskrivs i tabellen:
En form av spinal muskelatrofi |
Symptomatologi |
Spinal muskelatrofi typ I (barnet kan inte sitta upp, genomsnittlig livslängd - upp till 2 år) |
Det manifesterar sig från födseln till sex månader. Otillräcklig muskelton upptäcks, ropet är svagt, muskelsvaghet (inklusive tugga och svälja muskler) ökar. Det finns problem med huvudretention, barnet antar en "groda" hållning när man ligger ner. |
Spinal muskelatrofi typ II (barnet kan sitta upp, livslängden är vanligtvis mer än 2 år och mer än hälften av patienterna lever för att vara 20-25 år) |
Den debuterar med början vid 7 månaders ålder och upp till en och ett halvt års ålder. Svälja, andnings- och hostproblem märks ibland. Permanenta tecken inkluderar muskelspasmer, begränsad ledmobilitet, krökning av ryggraden, lågt blodtryck och muskelsvaghet. |
Spinal muskelatrofi typ III (barnet kan sitta och röra sig, men ovanstående förmågor går gradvis förlorade, livslängden är normal) |
Debuterar vid ett och ett halvt års ålder. Krökning av ryggraden och bröstkorgen, muskelatrofi av bäcken och proximala ben och ökad ledmobilitet noteras. Att svälja är svårt. |
Spinal muskelatrofi typ IV |
Hänvisar till vuxenformen. Symtomatologi har mycket gemensamt med den för ryggradens muskelatrofi typ III. Svagheten ökar gradvis, skakningar och muskelfasciokulationer dyker upp med debuten vid 16-25 års ålder. |
Prognos
I Werdnig-Hoffman-syndromet är den genomsnittliga livslängden 1,5-2 år. Dödligt resultat i de flesta fall beror på ökande andningsfel och utveckling av inflammation i lungorna. Med snabbt andningsstöd i form av konstgjord ventilation är det möjligt att öka barnets förväntade livslängd. Det finns ett speciellt behov av kontinuerlig palliativ vård, som också krävs i spinal amyotrofi typ II. Patologier för de tredje och fjärde typerna kännetecknas av en mer gynnsam prognos.
Alla typer av spinal muskelatrofi är en allvarlig sjukdom. Alla familjemedlemmar till patienten kräver ständigt psykologiskt, informativt och socialt stöd. It is important for the patient to ensure adequate diagnosis and professional support from specialists such as pediatrician, neurologist, neurologist, pulmonologist, cardiologist, orthopedist, physiotherapist, etc. Despite the lack of specific therapy for the disease, symptomatic treatment is carried out, special nutrition is prescribed (both parenteral and enteral), various rehabilitation measures that contribute to slowing the progression of the pathology and prevent the emergence of komplikationer.
Många patienter beviljas ett funktionshinder och ett individuellt rehabiliteringssystem utarbetas.
Naturligt förekommande spinal muskelatrofi utan användning av specialutrustning för att stödja andning och utfodring i ungefär hälften av fallen slutar i döden av det sjuka barnet före två års ålder (mestadels typ I-sjukdom).