Medicinsk expert av artikeln

Nya publikationer
Polymyositis och dermatomyositis: orsaker, symtom, diagnos, behandling
Senast recenserade: 05.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
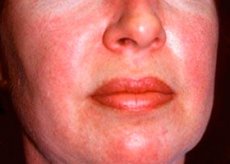
Polymyosit och dermatomyosit är sällsynta systemiska reumatiska sjukdomar som kännetecknas av inflammatoriska och degenerativa förändringar i muskler (polymyosit) eller muskler och hud (dermatomyosit). Den mest specifika hudmanifestationen är heliotroputslag.
Muskelengagemang är symmetriskt och inkluderar svaghet, viss ömhet och efterföljande atrofi av de proximala bäckengördelsmusklerna. Komplikationer kan inkludera visceralt engagemang och malignitet. Diagnosen baseras på klinisk presentation och bedömning av muskeldysfunktion genom att mäta enzymnivåer, utföra MR, elektromyografi och muskelbiopsi. Behandlingen innefattar glukokortikoider, ibland i kombination med immunsuppressiva medel eller intravenösa immunglobuliner.
Kvinnor blir sjuka dubbelt så ofta som män. Sjukdomen kan förekomma i alla åldrar, men upptäcks oftast i åldersintervallet 40 till 60 år; hos barn - från 5 till 15 år.
 [
[ Vad orsakar dermatomyosit och polymyosit?
Orsaken till sjukdomen tros vara en autoimmun reaktion mot muskelvävnad hos genetiskt predisponerade individer. Sjukdomen är vanligare vid förekomst av en belastad familjehistoria och bärare av vissa HLA-antigener (DR3, DR52, DR56). Möjliga utlösande faktorer är viral myosit och maligna neoplasmer. Det finns rapporter om upptäckt av strukturer som liknar picornavirus i muskelceller; dessutom kan virus orsaka liknande sjukdomar hos djur. Sambandet mellan maligna tumörer och dermatomyosit (mycket mindre frekvent än vid polymyosit) tyder på att tumörtillväxt också kan vara en utlösande faktor för sjukdomens utveckling som ett resultat av initiering av autoimmuna reaktioner mot vanliga antigener i tumören och muskelvävnaden.
Avlagringar av IgM, IgG och den tredje komponenten av komplement finns i väggarna i blodkärlen i skelettmusklerna; detta är särskilt karakteristiskt för dermatomyosit hos barn. Patienter med polymyosit kan också utveckla andra autoimmuna processer.
Patofysiologi för dermatomyosit och polymyosit
Patologiska förändringar inkluderar cellskador och atrofi mot bakgrund av inflammation av varierande svårighetsgrad. Musklerna i övre och nedre extremiteter, såväl som ansiktet, skadas i mindre utsträckning än andra skelettmuskler. Skador på viscerala muskler i svalget och övre matstrupen, mer sällan hjärtat, magsäcken eller tarmarna, kan leda till dysfunktion i ovanstående organ. Höga koncentrationer av myoglobin på grund av rabdomyolys kan orsaka njurskador. Inflammatoriska förändringar i leder och lungor kan också förekomma, särskilt hos patienter som har antikroppar mot antisyntetas.
Symtom på dermatomyosit och polymyosit
Polymyosit kan uppstå akut (särskilt hos barn) eller subakut (oftare hos vuxna). Akut virusinfektion föregår ibland eller är en utlösande faktor för sjukdomens manifestation, vars vanligaste manifestationer är svaghet i proximala muskler eller hudutslag. Smärta uttrycks i mindre utsträckning än svaghet. Polyartralgi, Raynauds fenomen, dysfagi, lungsjukdomar, allmänna symtom (ökad kroppstemperatur, minskad kroppsvikt, svaghet) kan utvecklas. Raynauds fenomen förekommer ofta hos patienter med samtidiga bindvävssjukdomar.
Muskelsvaghet kan fortskrida under flera veckor eller månader. För att muskelsvaghet ska visa sig kliniskt krävs dock att minst 50 % av muskelfibrerna är påverkade (alltså indikerar förekomsten av muskelsvaghet progression av myosit). Patienter kan uppleva svårigheter att lyfta armarna över axelhöjd, gå i trappor eller resa sig från sittande position. På grund av allvarlig svaghet i bäcken- och skuldergördelsmusklerna kan patienter vara bundna till rullstol eller säng. Om nackböjarna påverkas blir det omöjligt att lyfta huvudet från kudden. Påverkan på musklerna i svalget och övre matstrupen leder till sväljningssvårigheter och uppstötningar. Musklerna i nedre, övre extremiteter och ansiktet påverkas vanligtvis inte. Kontrakturer i extremiteterna kan dock utvecklas.
Hudutslag som observeras vid dermatomyosit är vanligtvis mörka i färgen och erytematösa. Periorbitalt ödem med lila färg (heliotroputslag) är också karakteristiskt. Hudutslagen kan vara något upphöjda över hudnivån och vara släta eller fjälliga; utslagen är lokaliserade i pannan, nacken, axlarna, bröstet, ryggen, underarmarna, de nedre delarna av smalbenen, ögonbrynen, knäområdet, mediala malleolerna, dorsala ytor av interfalangeala och metakarpofalangeala leder, på den laterala sidan (Gottrons symptom). Hyperemi vid naglarnas bas eller periferi är möjlig. Deskvamativ dermatit, åtföljd av sprickor, kan utvecklas på huden på fingrarnas laterala yta. Primära hudlesioner försvinner ofta utan följdsjukdomar, men kan leda till sekundära förändringar såsom mörk pigmentering, atrofi, ärrbildning eller vitiligo. Subkutana förkalkningar kan utvecklas, särskilt hos barn.
Ungefär 30 % av patienterna utvecklar polyartralgi eller polyartrit, ofta åtföljt av ödem och ledaus. Svårighetsgraden av ledmanifestationerna är dock liten. De uppträder oftare när antikroppar mot Jo-1 eller andra syntetaser detekteras hos patienterna.
Engagemang av inre organ (förutom svalget och övre matstrupen) är mindre vanligt vid polymyosit än vid andra reumatiska sjukdomar (särskilt SLE och systemisk skleros). I sällsynta fall, särskilt vid antisyntetassyndrom, manifesterar sig sjukdomen som interstitiell pneumonit (i form av dyspné och hosta). Hjärtarytmier och ledningsstörningar kan utvecklas, men de är vanligtvis asymptomatiska. Gastrointestinala manifestationer är vanligare hos barn som också har vaskulit och kan inkludera blodiga kräkningar, melena och tarmperforation.
Var gör det ont?
Klassificering av polymyosit
Det finns 5 typer av polymyosit.
- Primär idiopatisk polymyosit, som kan uppstå i alla åldrar, involverar inte huden.
- Primär idiopatisk dermatomyosit liknar primär idiopatisk polymyosit men involverar huden.
- Polymyosit och dermatomyosit i samband med maligna tumörer kan förekomma hos patienter i alla åldrar; deras utveckling observeras oftast hos äldre patienter, såväl som hos patienter med andra bindvävssjukdomar. Utvecklingen av maligna tumörer kan observeras både inom 2 år före och inom 2 år efter myositens debut.
- Polymyosit eller dermatomyosit hos barn är associerad med systemisk vaskulit.
- Polymyosit och dermatomyosit kan också förekomma hos patienter med andra bindvävssjukdomar, oftast progressiv systemisk skleros, blandad bindvävssjukdom och SLE.
Att inkludera myosit i bålens muskler i gruppen polymyosit är felaktigt, eftersom den senare är en separat sjukdom som kännetecknas av kliniska manifestationer som liknar dem vid kronisk idiopatisk polymyosit. Den utvecklas dock i ålderdom, drabbar ofta musklerna i kroppens distala delar (till exempel övre och nedre extremiteter), har en längre varaktighet, svarar mindre bra på behandling och kännetecknas av en typisk histologisk bild.
Diagnos av dermatomyosit och polymyosit
Polymyosit bör misstänkas hos patienter med besvär över proximal muskelsvaghet, med eller utan ömhet. Utredning för dermatomyosit är nödvändig hos patienter med besvär över utslag som liknar heliotrop eller Gottrons tecken, såväl som hos patienter med manifestationer av polymyosit i kombination med hudlesioner som överensstämmer med dermatomyosit. De kliniska manifestationerna av polymyosit och dermatomyosit kan likna de vid systemisk skleros eller, mer sällan, SLE eller vaskulit. Diagnossäkerheten ökar genom att så många som möjligt av följande fem kriterier uppfylls:
- svaghet i proximala muskler;
- karakteristiska hudutslag;
- ökad aktivitet av muskelvävnadsenzymer (kreatinkinas eller, i frånvaro av en ökning av dess aktivitet, aminotransferaser eller aldolas);
- karakteristiska förändringar i myografi eller MR;
- karakteristiska histologiska förändringar i en muskelvävnadsbiopsi (absolut kriterium).
Muskelbiopsi kan utesluta vissa kliniskt liknande tillstånd, såsom myosit i bålens muskler och rabdomyolys på grund av virusinfektion. Förändringar som avslöjas vid histologisk undersökning kan variera, men kronisk inflammation, fokus på muskeldegeneration och regeneration är typiska. En noggrann diagnos (vanligtvis genom histologisk verifiering) är nödvändig innan potentiellt toxisk behandling påbörjas. MR kan upptäcka fokus på ödem och inflammation i musklerna, följt av deras riktade biopsi.
Laboratoriestudier kan bekräfta eller tvärtom eliminera misstanken om sjukdomens förekomst, och är också användbara för att bedöma dess svårighetsgrad, möjligheten till en kombination med annan liknande patologi och diagnostisera komplikationer. Trots att antinukleära antikroppar detekteras hos vissa patienter är detta fenomen mer typiskt för andra bindvävssjukdomar. Cirka 60 % av patienterna har antikroppar mot cellkärnans (PM-1) eller hela tymuscellers antigen och Jo-1. Autoantikropparnas roll i sjukdomens patogenes är fortfarande oklar, även om det är känt att antikroppar mot Jo-1 är en specifik markör för antisyntetassyndrom, inklusive fibroserande alveolit, lungfibros, artrit och Raynauds fenomen.
Regelbunden bedömning av kreatinkinasaktivitet är användbar för att övervaka behandlingen. Hos patienter med svår muskelförtvining kan dock enzymaktiviteten vara normal trots förekomst av kronisk aktiv myosit. MR, muskelbiopsi eller förhöjd kreatinkinasaktivitet är ofta användbara för att skilja mellan återfall av polymyosit och glukokortikoidinducerad myopati.
Eftersom många patienter har odiagnostiserade maligniteter rekommenderar vissa författare screening av alla vuxna med dermatomyosit och de med polymyosit över 60 år enligt följande schema: fysisk undersökning, inklusive bröst-, bäcken- och rektalundersökningar (inklusive avföringstest för ockult blod); fullständig blodstatus; blodkemi; mammografi; karcinoembryonalt antigentest; urinanalys; lungröntgen. Vissa författare ifrågasätter behovet av sådan screening hos yngre patienter som inte har kliniska tecken på malignitet.
Vad behöver man undersöka?
Hur man undersöker?
Behandling av dermatomyosit och polymyosit
Tills inflammationen har lindrats bör fysisk aktivitet begränsas. Glukokortikoider är förstahandsbehandling. I sjukdomens akuta skede bör vuxna patienter förskrivas prednisolon (oralt) i en dos på 40 till 60 mg per dag. Regelbunden bestämning av kreatinkinasaktivitet är en tidig indikator på effektivitet: hos de flesta patienter noteras en minskning eller normalisering inom 6 till 12 veckor efter en ökning av muskelstyrkan. Efter normalisering av enzymaktiviteten minskas prednisolondosen: först med cirka 2,5 mg per dag under en vecka, sedan snabbare; om den ökade muskelenzymaktiviteten återfaller ökas hormondosen igen. Tillfrisknade patienter kan klara sig utan glukokortikoider, men oftast behöver vuxna patienter långvarig glukokortikoidbehandling (10-15 mg prednisolon per dag). Den initiala dosen prednisolon för barn är 30-60 mg/m² en gång per dag. Vid remission i > 1 år hos barn kan glukokortikoidbehandlingen avbrytas.
I vissa fall upplever patienter som får höga doser glukokortikoider en plötslig ökning av muskelsvaghet, vilket kan vara förknippat med utvecklingen av glukokortikoidmyopati.
Vid otillräckligt svar på glukokortikoidbehandling, såväl som vid utveckling av glukokortikoidmyopati eller andra komplikationer som kräver dosreduktion eller utsättning av prednisolon, bör immunsuppressiva medel (metotrexat, cyklofosfamid, azatioprin, ciklosporin) användas. Vissa patienter kan få endast metotrexat (vanligtvis i doser som överstiger de som används för RA-behandling) i mer än 5 år. Intravenösa immunglobuliner kan vara effektiva hos patienter som är refraktära mot läkemedelsbehandling, men deras användning ökar behandlingskostnaden.
Myosit i samband med primära och metastatiska tumörer, såväl som myosit i bålmusklerna, är vanligtvis mer refraktära mot glukokortikoidbehandling. Remission av myosit i samband med maligna tumörer är möjlig efter att tumören avlägsnats.
Vad är prognosen för dermatomyosit och polymyosit?
Långvarig remission (och även klinisk återhämtning) över 5 år observeras hos mer än hälften av de behandlade patienterna; hos barn är denna siffra högre. Återfall kan dock inträffa när som helst. Den totala femårsöverlevnaden är 75 %, högre hos barn. Dödsorsakerna hos vuxna är svår och progressiv muskelsvaghet, dysfagi, minskad näring, aspirationspneumoni eller andningssvikt på grund av lunginfektioner. Polymyosit är allvarligare och mer resistent mot behandling om det finns skador på hjärta och lungor. Död hos barn kan inträffa på grund av intestinal vaskulit. Den allmänna prognosen för sjukdomen bestäms också av förekomsten av maligna tumörer.