Medicinsk expert av artikeln

Nya publikationer
Aper-syndromet
Senast recenserade: 04.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.

 [
[ Orsaker Aper-syndromet
Utvecklingen av syndromet provoceras av en störning i strukturen hos en gen som ligger på kromosom 10. Denna gen är ansvarig för processen med fingerseparation, och dessutom för den snabba stängningen av kraniala suturer.
Dessutom anses infektionssjukdomar som drabbats under graviditeten (såsom röda hund eller tuberkulos, syfilis eller influensa, samt hjärnhinneinflammation) och moderns exponering för röntgenstrålning vara orsakerna till patologin. Oftast upptäcks ett sådant syndrom hos barn som föds av äldre föräldrar.
Patogenes
Aperts syndrom är ärftligt. Dess typ är autosomalt dominant (dvs. i en familj där en av de blivande föräldrarna är sjuk är sannolikheten att få ett barn med denna sjukdom 50-100%).
En unik mutation i fibroblasttillväxtfaktorreceptor 2 (FGFR2) resulterar i ett ökat antal progenitorceller som utvecklas längs den osteogena vägen. Detta resulterar i slutändan i ökad subperiosteal benmatrixbildning och för tidig ossifikation av kranialsuturerna under fosterutvecklingen. Ordningen och hastigheten för sutursmältningen avgör graden av deformitet och funktionsnedsättning. När suturmaterialet har läkt begränsas tillväxten av andra vävnader vinkelrätt mot denna sutur, och de sammansmälta benen fungerar som en enda benställning.
Det första genetiska beviset för att syndaktyli vid Aperts syndrom beror på en defekt i keratinocyttillväxtfaktorreceptorn (KGFR) var observationen av en korrelation mellan KGFR-uttryck i fibroblaster och svårighetsgraden av syndaktyli.
Amblyopi och strabismus är vanligare hos patienter med FGFR2 Ser252Trp-mutationen, och diskatrofi är vanligare hos patienter med FGFR2 Pro253Arg-mutationen. Patienter med FGR2 Ser252Trp-mutationer har en signifikant högre prevalens av synnedsättning jämfört med patienter med FGFR2 Pro253Arg-mutationen.
Symtom Aper-syndromet
Vissa manifestationer av sjukdomen är tydligt märkbara redan vid barnets födsel, eftersom de utvecklas inuti moderns livmoder. Bland de viktigaste symptomen på syndromet:
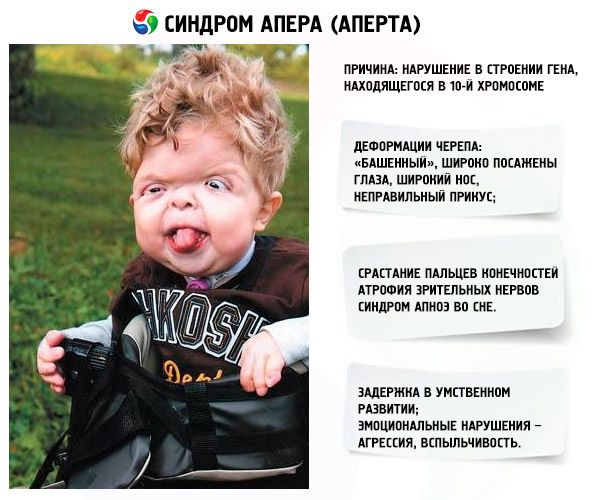
- Skallen är deformerad - den sträcker sig i höjd och får ett "tornliknande" utseende; dessutom är ögonen vida och lätt utbuktande (eftersom ögonhålornas storlek minskar); näsan vidgas och ett felaktigt bett bildas (de övre tänderna sticker ut för mycket);
- Fingrarna på extremiteterna är helt sammansmälta (främst ringfingret, såväl som långfingret och pekfingret) och ser ut som en hudhinna eller en fullständig sammansmältning av ben; dessutom kan extra fingrar växa;
- Psykisk utvecklingsstörning (förekommer inte hos alla);
- Synnerverna atrofierar, vilket leder till minskad synskärpa (i vissa fall uppstår fullständig synförlust);
- Ökat intrakraniellt tryck, som uppstår till följd av för tidig stängning av kraniala suturer, manifesterar sig i form av huvudvärk och kräkningar med illamående;
- Eftersom överkäken förblir underutvecklad uppstår andningsproblem;
- Sömnapnésyndrom är ett vanligt förekommande fenomen.
- Känslomässiga manifestationer – aggression, bristande återhållsamhet, stark irritabilitet.
Komplikationer och konsekvenser
Diagnostik Aper-syndromet
För att ställa en diagnos krävs följande steg:
- Läkaren bör analysera patientens klagomål, såväl som sjukdomshistorien. Det är nödvändigt att ta reda på om det fanns fall av liknande patologi i familjen;
- Neurologisk undersökning för att bedöma skallens form och patientens intellektuella utveckling (särskilda frågeformulär samt intervju);
- Undersökning av fundus för att upptäcka förekomsten av symtom på ökat intrakraniellt tryck (svullnad av synnerven, samt suddiga kanter);
- För att bedöma skallens tillstånd utförs en röntgenundersökning;
- Dator- och magnetisk resonanstomografi av huvudet för att undersöka hjärnans och skallens struktur lager för lager, för att fastställa förekomsten av symtom på för tidig sammansmältning av kraniala suturer, och dessutom hydrocefalus (på grund av ökat intrakraniellt tryck ackumuleras överskott av cerebrospinalvätska (detta är cerebrospinalvätskan som främjar den metaboliska processen, såväl som hjärnans näring));
- Röntgen av fötter och händer för att fastställa orsaken till fingrarnas sammansmältning (detta är viktigt för att planera efterföljande kirurgiska ingrepp);
- En konsultation med en medicinsk genetiker och en neurokirurg kan ordineras.
Tester
Genetisk analys av vanliga mutationer som förekommer i FGFR2-typgenen utförs.
Differentiell diagnos
Detta syndrom måste differentieras från andra genetiska patologier där kraniosynostos observeras. Det är sjukdomar som Pfeiffer-, Crouzons-, Saethre-Chotzen- och Carpenters syndrom. Molekylärgenetiska testmetoder används för att utesluta dessa anomalier.
Vem ska du kontakta?
Behandling Aper-syndromet
Kirurgi anses vara den enda effektiva behandlingen för Aperts syndrom – det hjälper till att korrigera individuella fysiska defekter och korrigerar även mental retardation.
Under denna procedur stängs den koronala suturen för att förhindra eventuell hjärnskada. Den vanligaste tekniken är kraniofacial distraktion, vilket innebär graderad skalltraktion. Ortodontisk och/eller ortognatisk kirurgi utförs för att avlägsna enskilda ansiktsdefekter.
Dessutom genomgår patienter kirurgisk korrigering av fingerfusionen.
Referenser
Medicinsk genetik. Nationell guide. Redigerad av Ginter EK, Puzyrev VP GEOTAR-Media. 2022