Medicinsk expert av artikeln

Nya publikationer
Amyloidos i levern
Senast recenserade: 29.06.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.
Amyloidos är vanligtvis en systemisk, generell patologi som kännetecknas av ansamling av amyloid (ett specifikt glykoprotein) i vävnader och efterföljande störningar av normal organfunktion. Leveramyloidos är mycket mindre vanligt än njure och mjälte [ 1 ] men åtföljer nästan alltid systemiska skador på kroppen. Ingen av de befintliga avbildningsteknikerna kan specifikt påvisa förekomsten av amyloid. Även vid klinisk och radiologisk misstanke är diagnosen amyloidos beroende av vävnadsbiopsi för att bekräfta förekomsten av amyloidavlagringar. [ 3 ] Behandlingen är komplex, omfattande och inkluderar immunsuppressiva och symtomatiska åtgärder. I svåra fall kan levertransplantation krävas.
Epidemiologi
Behandlingens framgång beror direkt på att sjukdomen, som orsakar bildandet av ett protein-polysackaridkomplex (amyloid) i olika organ och levern, ställs i rätt tid. Som praxis visar är amyloidos svår att anta eller misstänka, även om det är möjligt att identifiera och bekräfta den. Faktum är att i mer än 80 % av okända fall maskeras sjukdomen kliniskt av leverpatologi. Den mest effektiva diagnostiska metoden är biopsi.
Leveramyloidos är ett mer sällsynt problem jämfört med renal amyloidos. Samtidigt åtföljs alla fall av leverskador av skador på andra organ. Oftast drabbar patologin huvudsakligen strukturella delar av levertriaden, vilket avgör symtomatologins minimala och ospecificitet. Den kliniska och morfologiska bilden av hepatocellulär defekt och portalhypertension manifesteras i diffus och intralobulär typ av patologi.
En leverbiopsi är motiverad när hepatomegali föreligger utan tidigare leversymtom och i frånvaro av nefrotiskt syndrom.
Diffust leverengagemang ses i cirka 25 % av fallen, och hos 75 % av patienterna är endast portalvägarna drabbade.
Primär amyloidos drabbar levern i 90 % av fallen, medan sekundär amyloidos drabbar levern endast i 47 % av fallen.
Isolerat leverpåverkan är extremt sällsynt. Njurar (cirka 93 % av fallen), mjälte (72 %), hjärta (57 %), bukspottkörtel (36 %), binjurar (29 %), tarmar och lungor (21 % vardera) påverkas vanligtvis samtidigt.
Kvinnor får sjukdomen nästan dubbelt så ofta som män. Den genomsnittliga livslängden för amyloidospatienter är 52–64 år.
Orsaker hepatisk amyloidos
Amyloidos uppstår genom bildandet och ackumuleringen av ett komplext polysackarid-proteinkomplex - amyloid - i levervävnaden. Problemet med uppkomsten av den primära lesionen är hittills otillräckligt studerat. När det gäller sekundär patologi är dess uppkomst vanligtvis förknippad med sådana sjukdomar:
- Kroniska infektionsprocesser (tuberkulos, syfilis, aktinomykos);
- Purulenta inflammatoriska processer (mikrobiell endokardit, osteomyelit, bronkiektatisk sjukdom, etc.);
- Maligna sjukdomar (leukemi, visceral cancer, lymfogranulomatos).
Den reaktiva formen av amyloidos förekommer hos patienter med samtidig ateroskleros, reumatologiska sjukdomar (Bechterews sjukdom, reumatoid artrit), psoriasis, kroniska inflammatoriska och multisystemprocesser (inklusive sarkoidos). De viktigaste riskfaktorerna: ärftlig predisposition, cellulära immunitetsstörningar, hyperglobulinemi.
Patogenes
Det finns ett antal antaganden om ursprunget till leveramyloidos. De flesta specialister håller sig till versionen av dysproteinos, sjukdomens immunologiska och mutationella natur, såväl som lokal cellulär genes. Versionen av cellulär genes inkluderar förändringar i reaktioner som verkar på cellnivå (bildning av fibrillära prekursorer till amyloid av ett komplex av makrofager), även om amyloid bildas och ackumuleras utanför cellstrukturerna.
Versionen av dysproteinos baseras på det faktum att amyloid är en produkt av felaktig proteinmetabolism. Den grundläggande patogenetiska länken till problemet ligger i dysproteinemi och hyperfibrinogenemi, vilket leder till ansamling av grovt dispergerade protein- och paraproteinfraktioner i plasman.
Enligt den immunologiska versionen orsakas amyloidbildning av en antigen-antikroppsreaktion, där vävnadsnedbrytningsprodukter eller främmande proteiner fungerar som antigener. Amyloidansamling förekommer huvudsakligen i området med antikroppsbildning och överdriven närvaro av antigener.
Den mest troliga versionen anser forskare att mutationsteorin är, som tar hänsyn till en mängd olika mutagena faktorer som kan leda till avvikelser i proteinsyntesen.
Amyloid är ett komplext hypoprotein som består av globulära och fibrillära proteiner i kombination med polysackarider. Amyloidansamlingar påverkar intima och adventitia i kärlnätet, stroma i parenkymorgan, körtlarnas struktur etc. Amyloidansamlingar orsakar inte funktionella skador. Små ansamlingar orsakar inte funktionella störningar, men vid intensiv amyloidnärvaro i levern ökar volymen, organets utseende förändras och funktionsnedsättning utvecklas.
Leveramyloidos kännetecknas av avsättning av amyloidfibriller i Dysses utrymme, vilket vanligtvis börjar i den periportala regionen, även om det ibland är centrilobulärt och även kan avsättas i leverkärlen. [ 4 ], [ 5 ] I svåra fall leder amyloidavsättning till tryckatrofi av hepatocyter, vilket förhindrar passage av galla, vilket resulterar i kolestas, eller kan blockera sinusoiderna, vilket resulterar i portalhypertension. [ 6 ], [ 7 ], [ 8 ]
Symtom hepatisk amyloidos
Den kliniska bilden vid leveramyloidos är varierande och beror på intensiteten av amyloidackumulering, dess biokemiska egenskaper, den patologiska processens varaktighet, graden av organskada och kränkningen av deras funktionella tillstånd.
I det latenta stadiet av amyloidos, när amyloidansamlingar i levern endast kan detekteras genom mikroskopisk undersökning, saknas de första tecknen på sjukdomen. Med vidare utveckling och ökande funktionsunderskott i organet fortskrider symtomatologin.
Levern förtjocknar gradvis och förstoras. Palpationsmetoden kan palperas med förändrad, men organets gränser är släta och smärtfria. I sällsynta fall åtföljs patologin av smärta i det subkostala området på höger sida, dyspepsi, förstorad mjälte, gulfärgning av hud, slemhinnor och senhinnor, hemorragiskt syndrom.
De mest karakteristiska symtomen vid leveramyloidos: [ 9 ], [ 10 ]
- Amyloidackumulering i levern orsakar hepatomegali hos 33–92 % av patienterna;
- Mild gulsot
- Portal hypertension;
- Måttlig till svår kolestas.
Eftersom amyloidos mycket sällan drabbar endast ett organ, finns det vanligtvis ytterligare symtom:
- När njurskador utvecklas nefrotiskt syndrom och arteriell hypertoni med ytterligare njursvikt, ödem, ibland njurvenetrombos, leukocyturi, hematuri, hypoproteinemi, azotemi och så vidare;
- När hjärtat påverkas utvecklas ett tillstånd som liknar restriktiv kardiomyopati (rytmrubbningar, kardiomegali, ökande hjärtsvikt, svaghet och dyspné, ödem, mindre ofta - vätskeansamling i buk- och pleurahålan, perikardit);
- Om matsmältningskanalen påverkas kan makroglossi, svaghet och peristaltik i matstrupen, illamående och halsbränna, förstoppning eller diarré etc. förekomma;
- När bukspottkörteln påverkas uppstår symtomen på kronisk pankreatit;
- Om den muskuloskeletala mekanismen är involverad utvecklas symmetrisk polyartrit, karpaltunnelsyndrom och myopatier, och om nervsystemet påverkas upptäcks polyneuropatier, förlamning, ortostatiskt lågt blodtryck, ökad svettning och demens.
Om den patologiska reaktionen sprider sig till huden, uppstår många vaxartade plack i ansiktet, på halsen och i hudvecken. Bilden av neurodermatit, skivepitelfeber och sklerodermi är möjlig.
Kombinationen av flera amyloidlesioner och de olika symtomen gör det mycket svårare att identifiera hepatisk amyloidos och kräver en omfattande och fullständig diagnos.
Formulär
Enligt WHO:s klassificering skiljer man mellan fem typer av amyloidos:
- AL (primär);
- AA (sekundär);
- ATTR (ärftlig och senil systemisk);
- Aβ2M (hos patienter i hemodialys);
- AIAPP (hos patienter med insulinoberoende diabetes mellitus);
- AB (för Alzheimers sjukdom);
- AANF (senil förmaksamyloidos).
Det finns en lokal amyloidos i levern, men oftare är det en systemisk lesion, där den patologiska processen även involverar njurar, hjärta, mjälte, nervsystem, såväl som andra organ och vävnader.
Komplikationer och konsekvenser
Systemisk amyloidos leder gradvis till utvecklingen av akuta patologiska processer som i sin tur kan leda till döden. Bland de vanligaste och livshotande komplikationerna är följande:
- Frekventa infektiösa (bakteriella, virala) patologier, inklusive lunginflammation, pyelonefrit, glomerulonefrit;
- Kronisk lever- och njursvikt;
- Kronisk hjärtsvikt (kan föregå hjärtinfarkt);
- Hemorragiska stroke.
Venös trombos uppstår som ett resultat av ansamling och avsättning av proteiner på venväggarna. Lumen i de drabbade kärlen smalnar av, organsvikt utvecklas. Med tiden, mot bakgrund av långvarig hyperproteinemi, kan kärlet helt stängas igen. Alla komplikationer kan leda till ett ogynnsamt resultat - döden.
Diagnostik hepatisk amyloidos
Vid misstanke om leveramyloidos utförs diagnostiska åtgärder efter obligatoriska konsultationer med både gastroenterolog och terapeut, samt reumatolog, kardiolog, hudläkare, neurolog, urolog. Det är viktigt att göra en omfattande utvärdering av anamnesdata och kliniska manifestationer, samt att genomföra en omfattande laboratorie- och instrumentdiagnostik.
Testerna inkluderar nödvändigtvis urin- och blodprov. Vid hepatisk amyloidos observeras ofta en kombination av leukocyturi med proteinuri och cylindruri, och hypoproteinemi - med hyperlipidemi, anemi, hyponatremi och hypokalcemi, minskat antal trombocyter. Paraproteiner detekteras i urin och serumelektrofores.
Instrumentell diagnostik inkluderar:
- EKG, Eko;
- Ultraljud under buk;
- Röntgen av magsäcken, matstrupen;
- Irrigografi, bariumröntgenstrålar;
- Endoskopi.
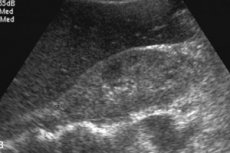
Radiologiska fynd vid hepatisk amyloidos inkluderar ospecifik hepatomegali, ökad ekogenicitet vid ultraljud eller densitet vid datortomografi (CT) och ökad T1-signalintensitet vid magnetisk resonanstomografi (MRI). [ 12 ] Scintigrafi med Tc-99m-relaterade indikatorer visar heterogent upptag, men det är ospecifikt. [ 13 ], [ 14 ] GC har visat sig öka leverstyvheten mätt med elastografi; [ 15 ], [ 16 ], [ 17 ] men det finns få fallrapporter. Magnetisk resonanselastografi (MRE) är för närvarande den mest exakta icke-invasiva metoden för att detektera och stadiebedöma leverfibros, [ 18 ], [ 19 ] MRE är användbart för att detektera progression, respons på behandling och förutsäga leverdekompensation hos patienter med leverfibros. [ 20 ]
Leverns amyloidos på ultraljud är svår att fastställa: en organförstoring bestäms, där den mest specifika hepatomegali överstiger 15 cm. Under ultraljudskontroll utförs en biopsi, vilket blir en avgörande indikator för diagnos. Med hjälp av en speciell nål tas en liten mängd levervävnad, sedan färgas den med ett speciellt färgämne och undersöks under ett mikroskop, vilket gör att man direkt kan se amyloidavlagringar.
En definitiv diagnos ställs först efter att amyloidfibriller har upptäckts i leverns och andra organs vävnad. Den genetiskt betingade typen av amyloidos bestäms genom noggrann genetisk-medicinsk undersökning av stamtavlan.
Differentiell diagnos
Amyloidos bör misstänkas hos alla patienter med en kombination av renal proteinuri, restriktiv kardiomyopati, autonom eller perifer neuropati och hepatomyeli, även i frånvaro av monoklonalt paraprotein. Att verifiera typen av amyloidos är mycket viktigt eftersom behandlingen av lesioner av olika etiologier är mycket olika.
Histologisk diagnos innebär färgning med Kongorött följt av mikroskopisk undersökning i polariserande ljus. Det är lämpligt att ta biopsi av flera vävnadsprover samtidigt. Om resultatet av färgningen blir positivt utförs immunhistokemisk analys med monoklonala antikroppar mot prekursorproteiner för att identifiera typen av amyloid.
DNA-analys utförs för att skilja mellan primär amyloidos och olika variationer av genetiskt betingad amyloidos. Amyloidfibriller kan isoleras från biopsiprover och sekvestreras till individuella aminosyror.
Ytterligare studier för att fastställa plasmacellsdyskrasi:
- Elektrofores av serumproteiner i blod och urin;
- Immunanalys för fria lätta kedjor;
- Immunfixering (immunoblotting) av serumproteiner;
- Benmärgsaspiration och trepanobiopsi.
Diagnos av leveramyloidos är en lång och arbetsintensiv process som kräver ökad uppmärksamhet från specialister och högkvalitativ utrustning från kliniker och laboratorier.
Vem ska du kontakta?
Behandling hepatisk amyloidos
Behandlingsåtgärderna syftar till att minska koncentrationen av redan existerande amyloidproteiner i blodet (eliminera orsaken till amyloidos) och stödja tillräcklig leverfunktion.
Sekundär amyloidos kräver blockering av den inflammatoriska processen (vid kroniska infektiösa och autoimmuna patologier). Vid autoimmuna sjukdomar rekommenderas användning av cytostatika. För att eliminera kroniska infektiösa processer avlägsnas ofta det inflammerade området kirurgiskt. Ofta kan denna metod stoppa ytterligare progression av amyloidos och förbättra leverfunktionen.
Primär amyloidos kräver användning av kemopreventiva läkemedel och ibland benmärgstransplantation.
Nuvarande riktlinjer rekommenderar kombinationen av cyklofosfamid, bortezomib, dexametason (CyBorD) och daratumumab som första linjens behandling för patienter som nyligen diagnostiserats med AL.
Bortezomib är en proteasomhämmare. Proteasomer är involverade i att minska proteotoxicitet och reglera proteiner som kontrollerar cellprogression och apoptos. Plasmaceller som genererar amyloid är särskilt känsliga för proteasomhämning eftersom de är beroende av proteasomen för att minska de toxiska effekterna av lätta kedjor och förhindra apoptos.
Daratumumumab är en monoklonal antikropp (mAb) som binder till CD38, ett transmembranglykoprotein som uttrycks på ytan av plasmaceller, vilket inducerar apoptos. Det är det enda läkemedlet som är specifikt godkänt för behandling av AL-amyloidos när det används tillsammans med CyBorD. Effekten av CyBorD-daratumumumab är mycket hög, där 78 % av patienterna uppnår ett signifikant hematologiskt svar (definierat som ett komplett svar eller mycket gott partiellt svar). Medianöverlevnaden i den lilla gruppen patienter som fick CyBorD (n = 15) var 655 dagar jämfört med 178 dagar för patienter som fick annan melfalan-dexametason-baserad behandling (n = 10).4
Dessa behandlingar har dock många biverkningar, inklusive kardiotoxicitet, vilket leder till behovet av dosreduktion eller avbrott i behandlingen, och användning av andra mindre effektiva men mer tolererbara terapeutiska strategier.
Isatuximab, en monoklonal antikropp mot CD38 liknande daratumumab, studeras för behandling av plasmacellsdyskrasi som ligger till grund för AL.
Tre monoklonala antikroppar, birtamimab, CAEL-101 och AT-03, studeras för närvarande för att avlägsna amyloidfibriller från sjuka organ. Resultaten av dessa studier kommer att kunna ge direkta bevis för hypotesen att genom att avlägsna lättkedjedeponeringsfibriller från organ sker en förbättring av organfunktionen. [ 21 ]
För att stödja leverfunktionen förskrivs läkemedel baserade på urso-deoxikolsyra (exempel - Ursosan). Urso-deoxikolsyra hjälper till att stabilisera cellmembran, minskar den negativa effekten av giftiga fettsyror i gallstasis som framkallas av amyloidavlagringar och hjälper till att återställa normalt gallflöde.
Dessutom symtomatisk behandling och stöd för funktionen hos andra vitala strukturer såsom nervsystem, hjärta, njurar etc. Stödjande behandling för patienter med hepatisk amyloidos inkluderar olika kliniska aspekter, inklusive behandling av hjärtsvikt, arytmier, ledningsstörningar, tromboembolism och samtidig förekomst av aortastenos.
Andra behandlingar beror på typen av amyloidos och vilka delar av kroppen som drabbas. Behandlingar kan inkludera: [ 22 ]
- Läkemedel som lindrar symtom, såsom smärtstillande medel, läkemedel mot illamående eller läkemedel som minskar svullnad (diuretika);
- Läkemedel för att minska amyloid;
- Njurdialys;
- Levertransplantation.
Levern producerar 95 % av TTR (transthyretin, ett protein involverat i tyroxin (T4)-transport och retinolbindande protein. Transthyretin syntetiseras huvudsakligen i levern och är rikt på beta-strängar som tenderar att aggregera till olösliga amyloidfibriller) mätt i serum. Därför har levertransplantation historiskt (sedan 1990) föreslagits som förstahandsbehandling för att eliminera den huvudsakliga källan till amyloidogen TTR hos patienter med den familjära formen (ATTRv), medan det inte är indicerat för ATTR-wt-formen. Levertransplantation av unga patienter i tidiga stadier av sjukdomen är associerad med en hög 20-årsöverlevnad. Levertransplantation verkar vara mer effektiv vid vissa mutationer och mindre effektiv vid andra, såsom V122I (associerad med kardiomyopati). Kombinerad lever- och hjärttransplantation är också möjlig hos unga ATTRv-patienter med kardiomyopati, och litteraturdata om en liten grupp patienter tyder på att denna kombination har en bättre prognos än enbart hjärttransplantation.
Patienter med hepatisk amyloidos är kontraindicerade att ta hjärtglykosider och kalciumantagonister såsom Diltiazem eller Verapamil, vilka kan ansamlas i amyloid. ACE-hämmare och betablockerare används med försiktighet.
Vid ortostatisk hypotoni förskrivs mineralkortikoider eller glukokortikoider, med hänsyn till att de kan orsaka dekompensation av hjärtsvikt. Även det alfa-adrenomimetiska läkemedlet midodrin (Gutron) används med försiktighet.
Antikonvulsiva och antidepressiva medel är lämpliga vid neuropatier.
I vissa fall av leveramyloidos måste läkare överväga organtransplantation.
Förebyggande
På grund av bristande information om patogenesen för leveramyloidos kan specialister inte utveckla ett specifikt förebyggande medel för sjukdomen. Därför inriktas huvudsakligen på att snabbt upptäcka och behandla eventuella kroniska patologier som kan provocera utvecklingen av sjukdomen. Om det finns fall av amyloidos av någon lokalisering i familjen rekommenderas det att systematiskt besöka läkare för apoteksundersökningar.
I allmänhet reduceras förebyggande åtgärder till att i tid eliminera infektionssjukdomar, särskilt de som tenderar att övergå i en kronisk process. Det handlar om att förhindra utveckling av tuberkulos, lunginfektioner etc. Det är viktigt att i tid upptäcka och behandla streptokockinfektioner, som kan orsaka kroniska former av autoimmuna inflammatoriska processer. Vi talar om scarlatina, streptokocktonsillit etc.
Om patienten redan har en autoimmun sjukdom, bör han systematiskt rådfråga en läkare, observera patologins aktivitet, applicera nödvändiga läkemedel enligt läkarens ordination och justera doserna enligt indikationerna.
Prognos
Prognosen för patienter med hepatisk amyloidos är ogynnsam. Sjukdomen förvärras långsamt men kontinuerligt, vilket så småningom orsakar dysfunktion i de drabbade organen och dödlig utgång – i synnerhet på grund av organsvikt.
Patienter med systemisk patologi dör huvudsakligen till följd av utvecklingen av kronisk njursvikt, även om hemodialys eller kontinuerlig ambulatorisk peritonealdialys i vissa fall förbättrar prognosen för sådana patienter. Överlevnadsgraden för patienter som får hemodialys, oavsett typ, kan jämföras med den för personer med andra systemiska patologier och diabetes mellitus.
Den främsta dödsorsaken under hemodialys är utvecklingen av komplikationer från hjärt-kärlsystemet.
Levertransplantation har länge ansetts vara en av de viktigaste behandlingsmetoderna för sjukdomen, och de mest optimistiska överlevnadssiffrorna observeras hos patienter vars ålder inte överstiger 50 år (förutsatt att den patologiska processen är kortlivad och BMI är normalt). Patienter med leveramyloidos i kombination med perifer neuropati har en något sämre prognos.