Nya publikationer
Nya rön bidrar till en bättre förståelse av orsakerna till Retts syndrom
Senast recenserade: 02.07.2025

Allt iLive-innehåll är mediekontrollerat eller faktiskt kontrollerat för att säkerställa så mycket faktuell noggrannhet som möjligt.
Vi har strikta sourcing riktlinjer och endast länk till välrenommerade media webbplatser, akademiska forskningsinstitut och, när det är möjligt, medicinsk peer granskad studier. Observera att siffrorna inom parentes ([1], [2] etc.) är klickbara länkar till dessa studier.
Om du anser att något av vårt innehåll är felaktigt, omodernt eller på annat sätt tveksamt, välj det och tryck på Ctrl + Enter.

Retts syndrom är en sällsynt neurologisk utvecklingsstörning för vilken det för närvarande inte finns något botemedel eller bra behandling. Det orsakar allvarliga fysiska och kognitiva symtom, av vilka många överlappar med autismspektrumstörningar.
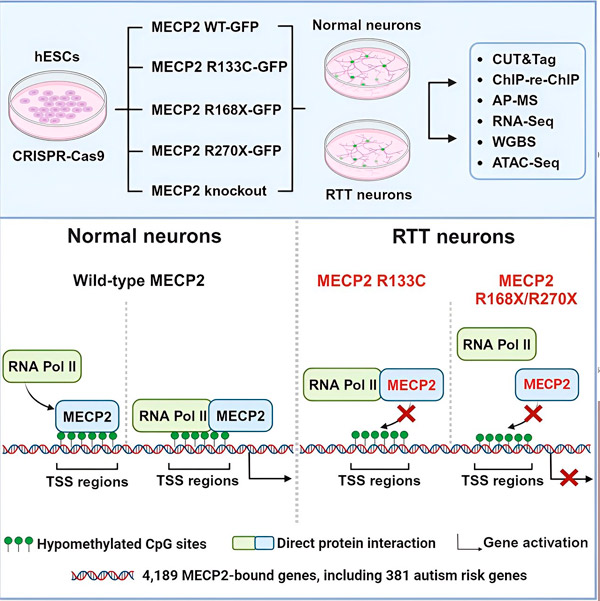
Retts syndrom orsakas av mutationer i MECP2-genen, som uttrycks i hög grad i hjärnan och verkar spela en viktig roll för att upprätthålla nervcellernas hälsa. Genen är belägen på X-kromosomen, och syndromet drabbar främst flickor. För att utveckla behandlingar för Retts syndrom vill forskare bättre förstå MECP2 och dess funktioner i hjärnan.
Forskare, inklusive Whitehead Institutes medgrundare Rudolf Jaenisch, har studerat MECP2 i årtionden, men många grundläggande fakta om genen förblev okända. Proteinet som kodas av genen, MECP2, är involverat i genreglering; det binder till DNA och påverkar uttrycksnivåerna av olika andra gener, eller mängden protein de producerar.
Forskarna hade dock inte en komplett lista över gener som påverkas av MECP2, och det fanns ingen enighet om hur MECP2 påverkar dessa gener.
Tidiga studier av MECP2 antydde att det var en repressor, vilket minskade uttrycket av dess målgener, men forskning av Jaenisch och andra hade tidigare visat att MECP2 också fungerar som en aktivator, vilket ökar uttrycket av dess mål – och att det kan vara en aktivator från första början. Okänd var också MECP2:s verkningsmekanism, eller exakt vad proteinet gör för att orsaka förändringar i genuttryck.
Teknologiska begränsningar har hindrat forskare från att få klarhet i dessa frågor. Men Yanish, hans postdoktor Yi Liu, och Yanishs tidigare laboratoriemedlem Anthony Flamier, nu biträdande professor vid forskningscentret CHU Sainte-Justine vid Université de Montréal, har använt banbrytande tekniker för att besvara dessa återstående frågor om MECP2 och få nya insikter i dess roll i hjärnhälsa och sjukdomar.
Deras resultat publicerades i tidskriften Neuron och forskarna skapade också ett online-arkiv för sina MECP2-data, MECP2-NeuroAtlas-portalen, som en resurs för andra forskare.
"Jag tror att den här artikeln fundamentalt kommer att förändra människors förståelse för hur MECP2 orsakar Retts syndrom. Vi har en helt ny förståelse för mekanismen, och den kan ge nya möjligheter att utveckla behandlingar för sjukdomen", säger Janisch, som också är professor i biologi vid MIT.
Djupare förståelse av MECP2 i hjärnan
Forskarna skapade först en detaljerad karta över var MECP2 binder i mänskliga neuronala gensekvenser, antingen inom gener eller i regulatoriska regioner av DNA nära dem. De använde en metod som kallas CUT&Tag, som kan lokalisera proteininteraktioner med DNA med hög precision.
Forskarna fann mer än 4 000 gener associerade med MECP2. De upprepade sin kartläggning i neuroner med vanliga MECP2-mutationer associerade med Retts syndrom för att avgöra var MECP2 är utarmat i sjukdomstillståndet.
Genom att veta vilka gener MECP2 binder till kunde Liu och Flamier börja koppla MECP2:s målproteiner till hjärnans hälsa. De fann att många av dess målproteiner är involverade i utvecklingen och funktionen av neuronala axoner och synapser.
De jämförde också sin lista över MECP2-mål med Simons Foundation Autism Research Initiative (SFARI) databas över autismassocierade gener och fann att 381 gener i den databasen är MECP2-mål.
Källa: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Dessa fynd kan bidra till att klargöra mekanismerna bakom autismsymtom vid Retts syndrom och ge en bra utgångspunkt för att undersöka MECP2:s möjliga roll vid autism.
"Vi har skapat den första integrerade kartan över MECP2-epigenomet inom hälsa och sjukdom, och den här kartan kan vägleda framtida forskning", säger Liu. "Att veta vilka gener som är måltavlor för MECP2, och vilka gener som är direkt störda i sjukdomen, ger en solid grund för att förstå Retts syndrom och ställa frågor om genreglering i neuroner."
Forskarna undersökte också om MECP2 ökade eller minskade uttrycket av dess målgener. I linje med historien om att MECP2 identifierats av vissa som en aktivator och av andra som en repressor, fann Liu och Flamier exempel där MECP2 spelade båda rollerna.
Men medan MECP2 oftare betraktas som en repressor, fann Liu och Flamier att det mestadels är en aktivator – vilket bekräftar tidigare fynd av Jaenisch och Liu. Ett nytt experiment visade att MECP2 aktiverar minst 80 % av sina mål, och ett annat fann att det aktiverar upp till 88 % av sina mål.
Kartan över målgener som forskarna skapade gav ytterligare insikt i MECP2:s roll som aktivator. De fann att för gener som MECP2 aktiverar binder den vanligtvis till en region av DNA uppströms genen som kallas transkriptionsstartstället.
Det är här cellulära maskineriet initierar processen att transkribera en gen till RNA, varefter RNA:t översätts till ett funktionellt protein, vilket är produkten av genuttryck. Närvaron av MECP2 vid transkriptionsstartstället, där genuttrycket börjar, överensstämmer med dess roll som en genaktivator.
Forskarna började sedan undersöka vilken roll MECP2 spelar i genaktivering. De tittade på vilka molekyler MECP2 binder till på denna plats, utöver DNA, och fann att MECP2 interagerar direkt med ett proteinkomplex som kallas RNA-polymeras II (RNA Pol II). RNA Pol II är en viktig cellulär maskin som transkriberar DNA till RNA. RNA Pol II kan inte hitta gener på egen hand, så det kräver en mängd olika kofaktorer, eller proteinkollaboratörer, för att hjälpa det att göra sitt jobb.
Forskarna föreslår att MECP2 fungerar som en sådan kofaktor, som hjälper RNA Pol II att initiera transkription vid gener där MECP2 binder. Strukturanalys av MECP2 har identifierat delar av molekylen som binder till RNA Pol II, och andra experiment har bekräftat att förlust av MECP2 minskar närvaron av RNA Pol II vid lämpliga transkriptionsstartställen, såväl som uttrycksnivåerna av målgener.
Detta tyder på att Retts syndrom kan orsakas av minskad transkription av gener som MECP2 riktar sig mot på grund av MECP2-mutationer som förhindrar att det binder till RNA Pol II eller binder till DNA. I överensstämmelse med denna idé är de vanligaste MECP2-mutationerna som är associerade med sjukdom trunkeringar: mutationer där en del av proteinet saknas, vilket kan förändra interaktionen mellan MECP2 och RNA Pol II.
Forskarna hoppas att deras resultat inte bara kommer att förändra vår förståelse av MECP2, utan att en djupare och bredare förståelse av hur MECP2 påverkar hjärnans utveckling och funktion kan leda till nya insikter som kommer att hjälpa personer med Retts syndrom och relaterade sjukdomar, inklusive autism.
”Det här projektet är ett utmärkt exempel på Janisch-laboratoriets samarbetsinriktade natur”, säger Flamier. ”Rudolf och jag hade ett specifikt problem relaterat till Retts syndrom, och jag hade erfarenhet av CUT&Tag-tekniken, som kunde lösa problemet. Genom diskussioner insåg vi att vi kunde kombinera våra ansträngningar, och nu har vi ett stort arkiv med information om MECP2 och dess kopplingar till sjukdomar.”